Article Text

Abstract
In the UK, when a young person dies suddenly, the coroner is responsible for establishing the cause of death. They will ask a consultant pathologist to carry out an autopsy in order to ascertain when, where and how that person died. Once the cause of death is established and is due to natural causes, the coroner can issue a death certificate. Importantly, the coroner is not particularly interested in the cause of death as long as it is due to natural causes, which avoids the need for an inquest (a public hearing about the death). However, if no identifiable cause is established at the initial autopsy, the coroner can refer the heart to a cardiac pathologist, since the cause of death is usually due to heart disease in most cases. Consultant histopathologists are responsible for the analysis of human tissue from both living individuals and the dead in order to make a diagnosis of disease. With recent advancements in the management protocols for routine autopsy practice and assessment following the sudden death of a young individual, this review describes the role of the consultant histopathologist in the event of a sudden death of a young athletic individual, together with the older middle-aged ‘weekend warrior’ athlete. It provides concise mechanisms for the main causes of sudden cardiac death (including coronary artery disease, cardiomyopathies, valve abnormalities, major vessel ruptures and electrical conduction abnormalities) based on detailed autopsy data from our specialised cardiac pathology laboratory. Finally, the review will discuss the role of the histopathologist in the event of a ‘negative’ autopsy.
- Cardiology
- Cardiovascular epidemiology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
The cardiovascular benefits of regular physical activity are well established. It appears that elite endurance as well as mixed-sports (aerobic and anaerobic) athletes survive longer than the general population.1 But even in 490 BC, Phidippides, who ran 26.2 miles from Marathon to Athens delivering the news of the Greek victory over the Persians, collapsed and died after this first marathon, highlighting the fact that fit people can die with exertion. Since then with modern media exposure, deaths in the athletic population are highly publicised. The majority of deaths in athletes are due to underlying cardiovascular disease, which is often not diagnosed during life. The investigation of these deaths in previously fit individuals calls for a detailed autopsy with emphasis placed upon the examination of the heart.2
This review describes the role of the consultant histopathologist in the event of a sudden death of a young athletic individual, together with the older middle-aged ‘weekend warrior’ athlete. It will also provide concise mechanisms for the main causes of sudden cardiac death (SCD) (including coronary artery disease (CAD), cardiomyopathies, valve abnormalities, major vessel ruptures and electrical conduction abnormalities) based on detailed autopsy data from our specialised cardiac pathology laboratory. Finally, the review will discuss the role of the histopathologist in the event of a ‘negative’ autopsy.
The role of the cardiac histopathologist
In the UK, when a young person dies suddenly, the role of the coroner is to ascertain when, where and how that person died. The coroner is not particularly interested in the cause of death as long as it is due to natural causes, which avoids a police and/or legal inquest. However, if no identifiable cause is established at the initial autopsy, the coroner can refer the heart to a cardiac histopathologist since the majority of sudden deaths are due to cardiac causes. Cardiac pathologists are responsible for the analysis of the hearts and making a specific diagnosis of the cause of deaths.
Epidemiology of sudden death
There is an important distinction for the mechanism of exercise-related sudden death, which is based upon the individual's age. SCDs in older athletes (>35 or 40 years) are predominately caused by atherosclerotic CAD. In contrast, SCDs in young athletes (<35 years of age) are associated with a number of inherited cardiac diseases, particularly the cardiomyopathies and ion channelopathies. In the USA, the highest recorded number of events in a single year was 76, with an average of 66 deaths per year (range 50–76) over 6 years: 29% occurred in black individuals, 54% in high-school students and 82% with physical exertion during competition/training. Males predominated, and the most common cardiovascular causes were hypertrophic cardiomyopathy (HCM) (36%) and congenital coronary artery anomalies (17%).3 This study is ,however, limited by being based upon media reporting and referral to Minnesota hypertrophy centre.
Another study from the USA in National Collegiate Athletic Association (NCAA) student-athletes during a 5-year period, reported 273 deaths and a total of 1 969 663 athlete participant-years. Of these 273 deaths, 187 (68%) were due to non-medical or traumatic causes, 80 (29%) to medical causes and 6 (2%) to unknown causes. Cardiovascular-related sudden death was the leading cause of death in 45 (56%) of 80 medical cases, and represented 75% of sudden deaths during exertion. The incidence of SCD was 1:43 770 participants per year. Among NCAA Division I male basketball players, the rate of SCD was 1:3100 per year. SCD is the leading medical cause of death and death during exercise in NCAA student-athletes. Current methods of data collection underestimate the risk of SCDs. Accurate assessment of SCD incidence is necessary to shape appropriate health policy decisions and develop effective strategies for prevention.4
In the Veneto region of Italy, 200 cases of sudden death in the young (≤35 years) were investigated with detailed cardiac pathological examination. Coronary atherosclerosis accounted for 23%, arrhythmogenic right ventricular cardiomyopathy (ARVC) for 12.5%, mitral valve prolapse for 10%, conduction system abnormalities for 10%, congenital coronary artery anomalies for 8.5%, myocarditis for 7.5%, HCM for 5.5%, aortic rupture for 5.5%, dilated cardiomyopathy (DCM) for 5%, non-atherosclerotic acquired CAD for 3.5%, postoperative congenital heart disease for 3%, aortic stenosis for 2%, pulmonary embolism for 2% and other causes for 2%. Cardiac arrest remained unexplained in 6% of the cases.5 Marathon and community data from the UK6 ,7 emphasised the high prevalence of CAD.
Data from our histopathology laboratory from a wide age group (118 cases), with detailed cardiac pathological examination of SCDs, documented 113 (96%) male deaths. The mean age of SCDs in this series was 27.9±12.5 years (range 7–59 years). Seventy-five per cent of all deaths occurred in subjects aged ≤35 years and one-third (33%) in child or adolescent athletes (<18 years old). The greatest number of deaths (n=20) occurred in the 16–20 years age group. The vast majority of SCDs occurred during or immediately after exercise (81%). Cardiomyopathy was the dominant cause of death (n=73, 62%) with left ventricular hypertrophy (LVH) as the most commonly identified macroscopic abnormality within this group (n=49, 41%). The majority of these cases were associated with myocyte hypertrophy (n=36) including fibrosis (n=9) but no myocyte disarray and classified as idiopathic LVH. The remaining 13 cases exhibited classic myocyte disarray, the historically histological hallmark of HCM. ARVC was the second most common diagnosis and was identified in 13%. This study also highlighted for the first time the morphologically normal heart; pointing to electrical abnormalities as well as other cardiac pathology8 (figure 1). In France, from 2005 to 2010, the incidence of sports-related sudden death was 4.6 cases per million population per year, with 6% of cases occurring in young competitive athletes. More than 90% of cases occurred in the context of recreational sports, with the age of subjects being relatively young (46±15 years), with a predominance of men (95%), but causes of death were not included in the study.9

Chart showing the relative frequency of cardiac disease in cohort of 118 cases from our laboratory. ARVC, arrhythmogenic right ventricular cardiomyopathy; CM, cardiomyopathy; HCM, hypertrophic cardiomyopathy; IF, idiopathic fibrosis; ILVH, idiopathic left ventricular hypertrophy. This figure is only reproduced in colour in the online version.
What is clear from the literature and from our laboratory is that only a minority have experienced antecedent symptoms suggestive of underlying cardiac disease, including syncope, presyncope, chest pain, palpitations or shortness of breath prior to death, thus screening athletes based upon a questionnaire has limited value.8
Cardiac causes
CAD and cardiomyopathies predominate. Valve abnormalities and rupture of the aorta must also be considered. However more recently, electrical conduction abnormalities of the heart, the channelopathies, have risen in prominence in the context of the negative autopsy. For interest, I have published on detailed examination of the heart at autopsy.2
Coronary mechanisms of sudden death
Coronary artery disease
Most sudden deaths in athletes over the age of 35 are due to coronary artery atheroma with significant blockage (>75% narrowing of main vessel, usually the left anterior descending coronary artery) of the coronary arteries.10 ,11 There may be a substrate for sudden death such as acute or chronic infarction, but the cardiac muscle may be normal so the presumed mode of death is a sudden cardiac arrhythmia due to increased demands made on the myocardium by exercise (demand ischaemia). Transient left ventricular (LV) contractile dysfunction is observed owing to postexercise stunning in patients with CAD. Plaque rupture, thrombosis, acute infarction and fibrosis need not be present to attribute coronary atheroma as the cause of the sudden death. Obviously if the individual is obese, smoked, had high blood pressure, diabetes, elevated abnormal lipids or had a strong family history of heart disease, he/she will be at increased risk. The typical case is the weekend footballer or person deciding to run the marathon as a keep-fit challenge in middle age. Acute exercise leads to a transient activation of the coagulation system, which promotes thrombosis.12 Yet, young age is not a deterrent to attributing death to coronary atheroma. My youngest CAD death was an 11-year-old who died on a cross-country run and had widespread diffuse atheroma in all coronary arteries, who had been totally asymptomatic until she dropped dead. She had a strong family history of hypercholesterolaemia. Ischaemic heart disease is not covered further in this review. However, I always urge pathologists to be conservative in attributing death to CAD, unless there is significant atheroma in the left main stem, left anterior descending coronary artery or right coronary artery (RCA). Significant atheroma in the diagonal/marginal branches or left circumflex is open to debate when there is no thrombosis or evidence of ischaemic damage in the ventricle. I always advise pathologists to take samples for histology of coronary artery lesions in young people because not all blocked coronary vessels are due to atheroma.
Non-atheromatous coronary causes of SCDs
There are many non-atheromatous abnormalities of the coronary arteries, which can cause sudden death. In our recent study of 50 sudden deaths, 12 of the patients (24%) died during or immediately after physical exertion. Twenty-four (48%) had anomalous coronary arteries; eight (16%) had coronary artery dissection; six (12%) had coronary artery vasculitis; and six (12%) had coronary artery spasm. Only 20 of the 50 patients (40%) were documented to have experienced cardiac symptoms such as syncope, chest pain on exertion or breathlessness before their SCD.13
Coronary artery spasm on exercise
Myocardial ischaemia may not only be caused by plaque rupture and subsequent coronary thrombosis, but may also be due to coronary vasospasm (in the microvasculature as well as in epicardial coronary segments). Exercise-induced ischaemia and/or a pathological exercise test result may not only be caused by severe coronary stenoses, but may also be due to a reduced coronary perfusion reserve, secondary to microvascular dysfunction. Pro-atherogenic cardiovascular risk factors are not only associated with atherosclerotic CAD, but also with the occurrence of a coronary vasomotility disorder. Coronary artery spasm is a controversial topic for pathologists, as it is impossible to detect at autopsy. It is presumed to be the cause of SCD if there is transmural myocardial ischaemic damage, usually in the anterolateral wall of the left ventricle (region of distribution of the left coronary artery (LCA)) with normal coronary arteries.14 The ischaemic damage in this regional area points to a regional arterial transient blockage/spasm. Thrombotic/embolic disease with lysis is a possibility in these cases, but there is no source for thrombosis or emboli found at autopsy.
Drugs and CAD
Androgenic-anabolic steroid (AAS) use is associated with SCDs, myocardial infarction, altered serum lipoproteins and cardiac hypertrophy in humans who habitually use these drugs. There are at least four hypothetical models of AAS-induced adverse cardiovascular effects: (1) an atherogenic model involving the effects of AAS on lipoprotein concentrations; (2) a thrombosis model involving the effects of AAS on clotting factors and platelets; (3) a vasospasm model involving the effects of AAS on the vascular nitric oxide system; and (4) a direct myocardial injury model involving the effects of AAS on individual myocardial cells.15 Systematic toxicology indicates that 3.1% of SCDs are cocaine related and are mainly due to LVH, small vessel disease and premature coronary artery atherosclerosis, with or without lumen thrombosis.16 Smoking of cannabis is also related to coronary thrombosis and infarction.17 Toxicology is thus essential in all cases of sudden death in athletes to rule out these drugs being related to atheroma, thrombosis, infarction or hypertrophy. Screening for these drugs can be difficult since standard diagnostic criteria for substance dependence, usually crafted for acutely intoxicating drugs, must be adapted slightly for cumulatively acting drugs such as AAS.
Anomalous coronary arteries
Congenital anomalies of the origins and course of the coronary arteries can be simple anatomical variants without an effect on coronary blood flow. Dangerous anomalies are one artery taking origin from the pulmonary trunk or both arteries taking origin in one aortic sinus with the anomalous artery having an intramural aortic component and crossing between the aorta and pulmonary trunk (figure 2A,B). Arteries crossing between the aorta and pulmonary trunk tend to induce spasm on exercise, and so there is always a risk of sudden death with exercise. Both of these anomalies may present as myocardial infarction or angina in infancy or early adult life. An anomalous LCA originating from the right coronary sinus is considered more dangerous than the RCA originating from the left coronary sinus.13 High take-off of the coronary artery (greater than 10 mm above the sinutubular junction) has been associated with SCDs. It is well established that there is normal anatomical variation in the location of coronary ostia at, below and above the sinutubular junction. Muriago et al18 proposed that a measurement of 120% or more of the depth of the sinus should be used as the criterion for pathological high take-off. It is the combination of high take-off, slit-like opening, associated with an intramural course and ischaemic damage in the ventricle that make this entity important in establishing a cause of sudden death.

(A) Both arteries taking origin in one aortic sinus. (B) The anomalous coronary artery crossing between the aorta and pulmonary trunk. This figure is only reproduced in colour in the online version.
Coronary artery dissection
Spontaneous dissection of the coronary arteries has been described largely in young adults, especially in postpartum women. It is a rare cause of SCDs but can be missed unless looked for carefully.19 The process is distinct from aortic dissection and starts as an intramural haematoma, which compresses the vessel lumen from outside and can lead to SCD or myocardial infarction (figure 3). This haematoma may rupture into the lumen and a dissection track is created. Cases with spontaneous coronary artery dissection occurring after intense exercise or exertion have been described in the literature.20 There is also, like coronary artery spasm, an association with drug use, especially cocaine. The pathogenesis is not clear. Previously it was considered a vasculitis because of the presence of adventitial inflammatory eosinophils and basophils, which are a response to the dissection itself and is not an arteritis.

Demonstrating an intramural haematoma, which compresses the vessel lumen from outside. Haematoxylin and Trichrome stain of the coronary artery. This figure is only reproduced in colour in the online version.
Myocardial bridging
Myocardial bridging is a congenital coronary anomaly but its clinical relevance is debated.21 It is a condition in which a band of cardiac muscle overlies the coronary artery along its course for a varying length. As a consequence, compression of the coronary artery lumen in systole, extending into diastole, is the main functional consequence of this anatomic abnormality. It has been reported that this anomaly is of clinical and pathological significance when it involves the left anterior coronary artery which has a long (20–30 mm) and deep (5 mm) intramyocardial course (figure 4). An association between coronary artery spasm and myocardial bridging has been reported, and also in association with HCM. So finding a significant muscle bridge over the left anterior coronary artery should alert the pathologist to this cardiomyopathy. However, in our laboratory it is a rare cause of sudden death, but our cases all occurred during running.13
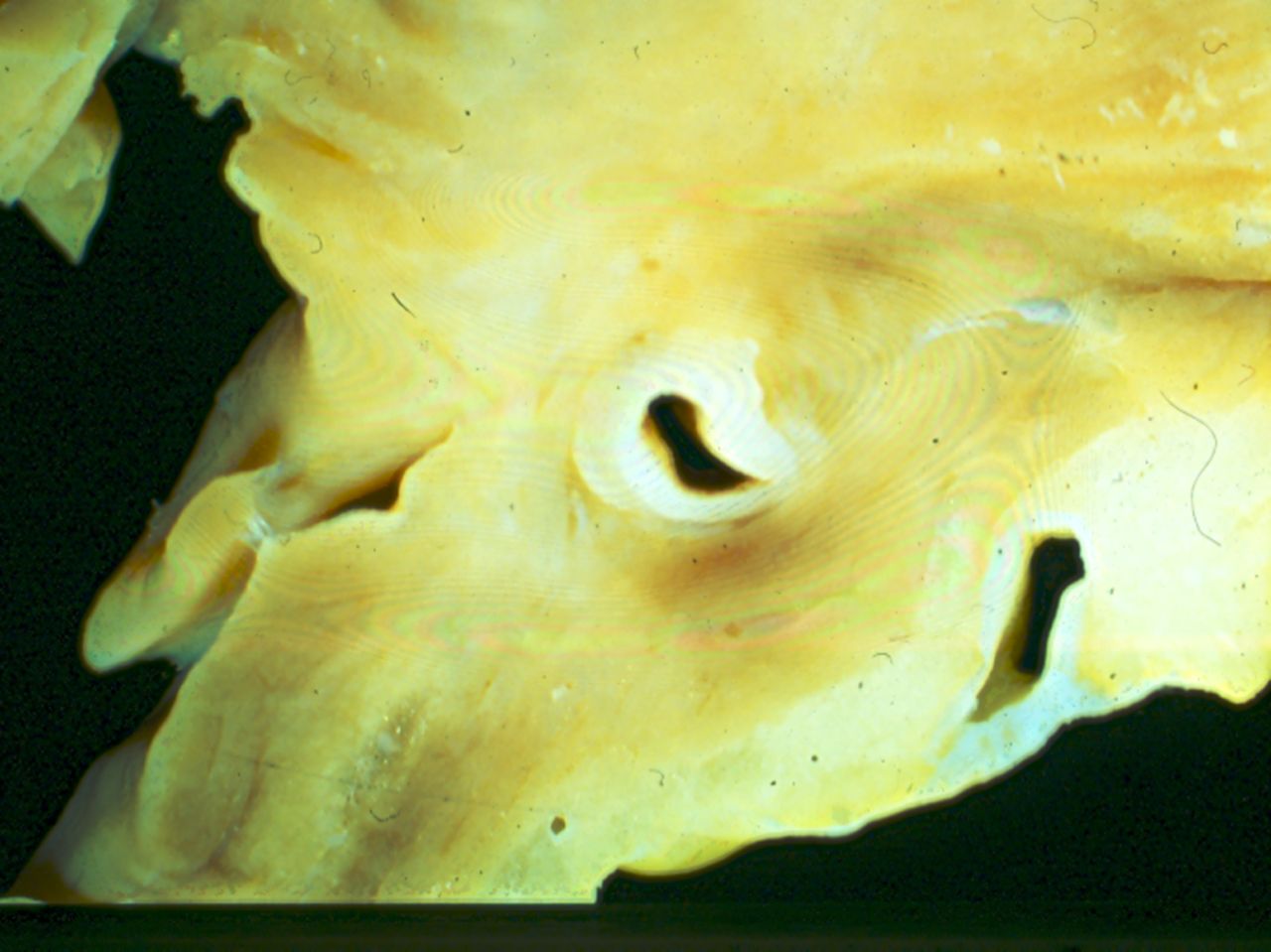
Coronary artery deep within muscle of the heart; left anterior coronary artery which has a long (20–30 mm) and deep (5 mm) intramyocardial course. This figure is only reproduced in colour in the online version.
Cardiomyopathic mechanisms of sudden death
Hypertrophic cardiomyopathy
HCM is inherited as a mendelian autosomal dominant trait and caused by mutations in any one of 11 genes, each encoding proteins of the cardiac sarcomere. It has an incidence of 1 in 500 of the general population and is considered to be one of the commonest forms of cardiac genetic disease. It is also disproportionately more prevalent in African-American athletes. Risk factors which increase the risk of sudden death include a family history of sudden death, extreme LV wall thickness (≥30 mm), non-sustained ventricular tachycardia, unexplained (non-neurocardiogenic) syncope particularly in young patients and inappropriate blood pressure response to exercise.22
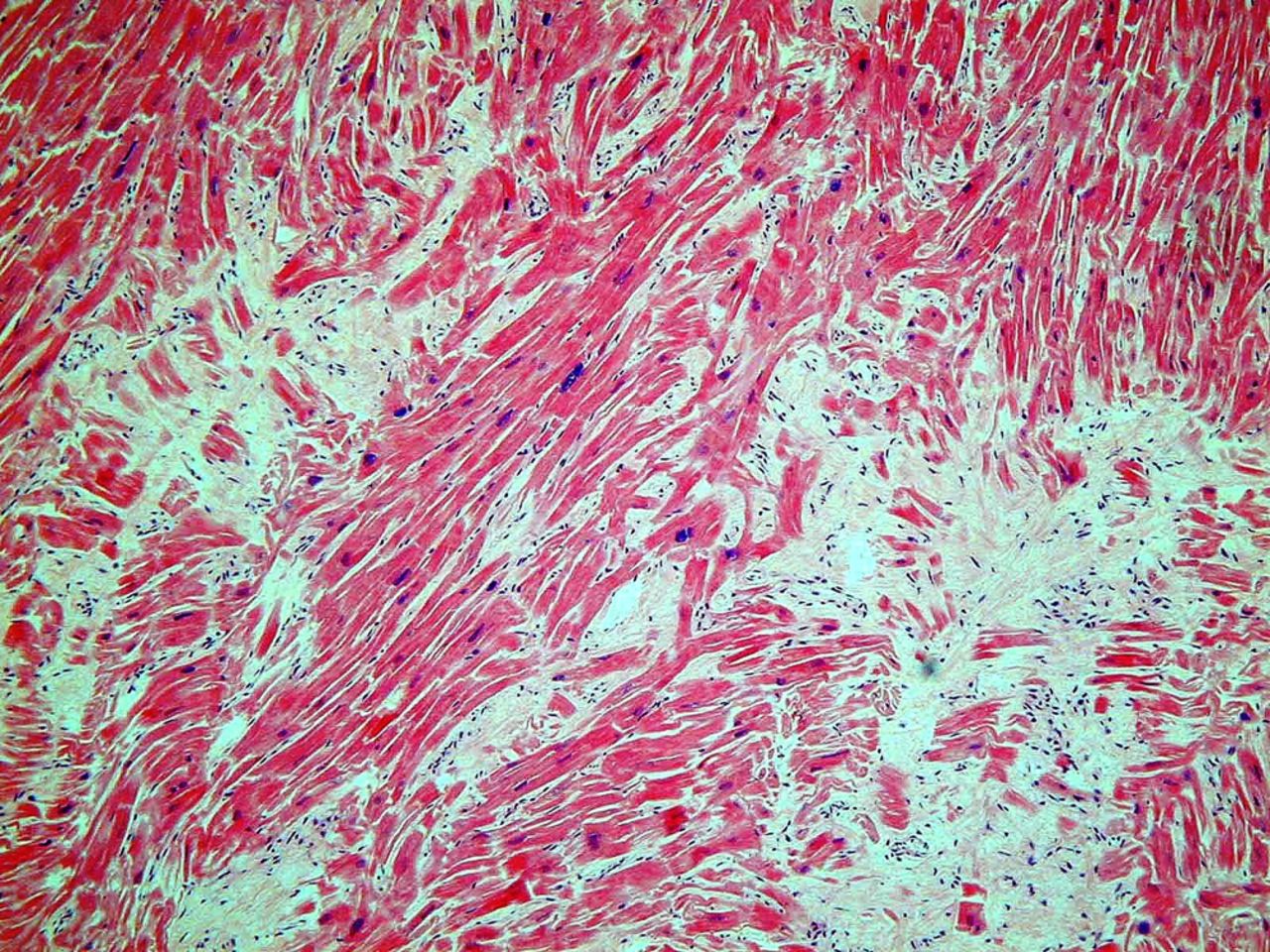
At autopsy, the heart weight exceeds 500 g with increase in LV wall thickness above 15 mm, with or without myocardial scarring (figure 5). In our experience, impact lesions on the left side of the upper interventricular septum due to imprint of the anterior mitral leaflet are only seen in a minority of cases. Also be aware of the fact that some cases of HCM may demonstrate normal heart weight and ventricular wall thickness, yet may present with microscopic myocyte disarray particularly in mutations in Troponin T. Myocyte cellular disarray can be widely distributed, occupying substantial portions of the LV wall, and is more extensive in young patients who die suddenly (figure 6).
Heart weighs 500 grams with increase in left ventricular wall thickness above 15 mm. This figure is only reproduced in colour in the online version.

Myocyte cellular disarray in the left ventricle. H&E staining of muscle. This figure is only reproduced in colour in the online version.
Myocyte disarray may not be related to the degree of hypertrophy and normal areas may have considerable disarray, while thickened areas show no histological features of disarray. The myocyte disarray can be focal, involving a small cluster of cells, or may involve large bundle disarray. The pathologist should be aware that focal myocyte disarray is always found in the anteroseptal and posteroseptal wall of the ventricles, at dividing points of trabeculae in the inner myocardium and around blood vessels, as well as around areas of scarring, so it is necessary to be cautious in diagnosing myocyte disarray unless it is obvious in most blocks examined in areas away from these zones, as well as areas of scarring. The interventricular septum is usually positive for this feature and the outer half of the free wall of the LV myocardium is another area that yields results free of artefact. I never make a diagnosis of HCM without significant myocyte disarray being present in at least 3 out of 10 blocks from the left ventricle. Myocyte disarray is common in the right ventricle (RV) because it has prominent trabeculae, so this should be avoided for diagnosis of HCM. LV scarring is prominent in most athletes who die suddenly with HCM and it is usually in the inner third of the LV. Consequently, using cardiac MRI for the identification of myocardial fibrosis has prognostic significance in identifying potential athletes with HCM.23 ,24
Idiopathic LVH
Athletic autopsy data from our laboratory demonstrates LVH without myocyte disarray to be the predominant finding (31%). Idiopathic LVH is becoming increasingly recognised and although it has been reported in previous studies, our data are the first series where it predominates. It is unclear as to whether idiopathic LVH is part of the HCM spectrum, a separate inherited entity or an acquired pathological variant of the physiological LVH, exhibited as part of the ‘athlete's heart’ syndrome, on certain predisposing genetic backgrounds. However, it should be noted that extreme LVH has sometimes been linked to the use of anabolic steroids, something that cannot be dismissed when undertaking an autopsy in an athlete that has died suddenly.25
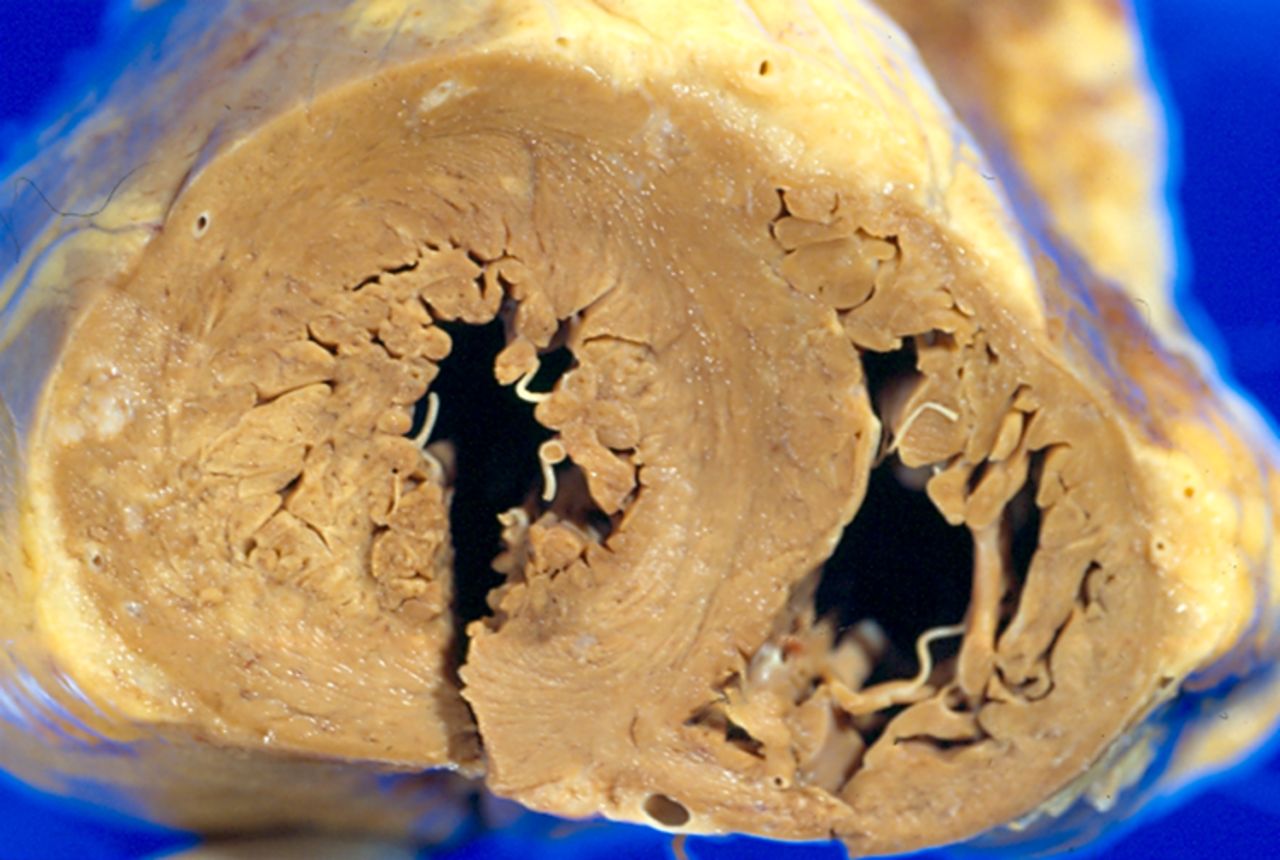
Arrhythmogenic right ventricular cardiomyopathy
In our laboratory, ARVC is the second most common cardiomyopathy accounting for 13% of the SCDs, with HCM being identified in an almost equal proportion (11%). The prevalence of ARVC in our cohort is comparable with data from other European countries which may represent a genetic predisposition in Caucasian individuals and more awareness of this entity within Europe compared to the USA. ARVC is the most recently described cardiomyopathy and, unlike other types, predominantly involves the RV. It is characterised by progressive replacement of RV myocardium by either segmental or diffuse fibrofatty tissue, often with the involvement of the LV wall. The LV is so frequently involved as to support the adoption of the broad term arrhythmogenic cardiomyopathy. Usually both ventricles are involved in the majority of cases. Rarely one gets involvement of the left ventricle alone and the distribution of the fat and fibrous tissue are on the epicardial surface. The fibrofatty replacement leads to ventricular arrhythmias, predisposing the athlete to potential SCD. A causative mutation in genes encoding desmosomal proteins has been identified, and thus ARVC is regarded as a genetically determined myocardial degenerative disease. There is a wide spectrum of macroscopic findings in ARVC ranging from normal through to dilated thin-walled appearance. Usually the RV shows dilatation and thinning of the wall with replacement by fat and fibrosis (figure 7). Usually the fat and scar tissue is in the epicardial aspect of the ventricle and extends in to replace the full thickness of the wall.26

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Wall of the right ventricle showing dilatation and thinning of the wall with replacement by fat and fibrous tissue. This figure is only reproduced in colour in the online version.
Dilated cardiomyopathy
DCM is an unusual cause of sudden death in athletes due to the fact that most patients with this entity are symptomatic and present with cardiac dysfunction; thus, engaging in sports that require high cardiac outputs would be unusual. In our laboratory, the range incidence ranges between 2% and 11%. However, DCM has a prevalence of 1 in 2500 individuals, arises more commonly in male adults than in women and is inherited in 20–48% of cases. Familial disease should always be suspected when there is a family history of premature cardiac death, or conduction system disease or skeletal myopathy. Autosomal-dominant forms of the disease are caused by mutations in cytoskeletal, sarcomeric protein/Z-band, nuclear membrane and intercalated disc protein genes. X linked diseases associated with DCM include muscular dystrophies (eg, Becker and Duchenne) and X linked DCM. DCM may also occur in patients with mitochondrial cytopathies and inherited metabolic disorders (eg, haemochromatosis). Examples of acquired causes of DCM include nutritional deficiencies, endocrine dysfunction and the administration of cardiotoxic drugs. At autopsy, the heart is heavy, weighing in excess of 500 g, with a thin-walled LV measuring less than 10 mm.
Stress cardiomyopathy
Takotsubo syndrome is a reversible neuromyocardial failure that has been thought to be related to an acute catecholamine toxicity of the myocardium brought upon by a stressful event. It is also known as apical ballooning syndrome and is a transient cardiomyopathy that mimics an acute myocardial infarction. The pathogenesis of this disorder is likely to be catecholamine mediated myocyte damage and microvascular dysfunction; however, a number of possible alternative theories have been suggested. These include oxidative stress, transient coronary obstruction and oestrogen deficiency, the last explaining the high prevalence of takotsubo cardiomyopathy in women. It can be triggered by hyponatraemia but is usually non-fatal.27 It has not surprisingly been reported in sport. High wall stress of the RV during intense sports may explain observed B-type natriuretic peptide elevations immediately after a race, may lead to cellular disruption and leaking of cardiac enzymes and may even result in transient RV dilatation and dysfunction. Over time, this could lead to chronic remodelling and a pro-arrhythmic state resembling ARVC in some cases. ARVC in high-endurance athletes may develop in the absence of underlying desmosomal abnormalities, probably only as a result of excessive RV wall stress during exercise. This has been labelled as ‘exercise-induced ARVC’.28
Valve abnormalities and major vessel mechanisms of sudden death
There is greater awareness of the bicuspid aortic valve (BAV) in the young population who practise sport. A normally functioning BAV usually does not represent a limit for practising sport. However, the stress of regular and intense exercise on an abnormal aortic valve may favour its early deterioration and accelerate the development of complications. Therefore, athletes with BAV warrant regular follow-up. While non-existent in elite athletes, I have cases of undiagnosed severe congenital aortic bicuspid stenosis where the patient has died while undertaking unaccustomed exercise, such as when they are being pursued or running for a train.
The relation between mitral valve prolapse and sudden death during exercise is much more complex.29 The natural history of floppy mitral valve is usually benign. A number of patients with floppy mitral valves have ventricular arrhythmias, in particular repeated ectopic beats; sudden death may occur in this subgroup but it is extremely rare; however, if it does, it usually occurs in young females.
Four inherited disorders known to effect major arteries include Marfan syndrome (MS), Ehlers-Danlos syndrome, BAV and non-syndromic familial aortic dissection. MS affects approximately 1 in 20 000 of the general population and is due to a mutation in the fibrillin gene, involved in elastin production of the connective tissues. People who have this medical condition are usually tall, slender and loose-jointed and may be volleyball/basketball players. Non-syndromic familial thoracic aortic aneurysms (TAADs) and dissections are inherited in families as an autosomal-dominant disorder due to mutations in collagen.30 Genes causing TAADs include TAAD1, TAAD2, FAA1 and ACTA2. These loci overlaps with a second locus for MS, termed the MFS2 locus.
Aortic rupture usually occurs during athletic activity. In our experience, acute aortic dissection can also occur at the time of intense physical exertion associated with transiently high LV afterloads, commonly seen within strength-trained athletes such as weightlifters, bodybuilders, throwers and wrestlers. Accordingly, a rapid rise in blood pressure, together with a personal history of hypertension is commonly associated with aortic dissection in this population of athletes.31
Chest trauma/impact mechanisms of sudden death
A blow to the chest in the area of the heart, called commotio cordis, or cardiac concussion is a rare cause of sudden death in athletes. This condition often occurs in children or adolescents with a non-penetrating and usually innocent appearing, blow to the middle of the chest, such as when a baseball, hockey puck, lacrosse ball, softball or karate blow strikes the athlete's chest. The precise incidence of commotio cordis is unknown because of the absence of systematic and mandatory reporting, but on the basis of data from the National Commotio Cordis Registry in Minneapolis, it is among the most frequent cardiovascular causes of sudden death in young US athletes, after HCM and congenital coronary artery anomalies.32 ,33 Since commotio cordis occurs in a wide variety of circumstances, it has undoubtedly been underreported. Most victims are boys or men and are white. Although cardiovascular collapse is virtually instantaneous, 20% of victims remain physically active for a few seconds after the blow (eg, continuing to walk, run, skate, throw a ball or even speak), which may reflect individual tolerance for sustained ventricular tachyarrhythmias. Commotio cordis is a primary arrhythmic event that occurs when the mechanical energy generated by a blow is confined to a small area of the precordium and profoundly alters the electrical stability of the myocardium, resulting in ventricular fibrillation. The first of these determinants involves the location of the blow, which must be directly over the heart (particularly at or near the centre of the cardiac silhouette). This finding is consistent with clinical observations at autopsy, that precordial bruises representing the imprint of a blow are frequently evident in victims. There is no evidence in humans or in experimental models that blows sustained outside the precordium (eg, the back, the flank or the right side of the chest) cause sudden death. The second determinant concerns the timing of the blow, which must occur within a narrow window of 10–20 ms on the upstroke of the T-wave, just before its peak (accounting for only 1% of the cardiac cycle)—that is, the blow must occur during an electrically vulnerable period, when inhomogeneous dispersion of repolarization is greatest, creating a susceptible myocardial substrate for provoked ventricular fibrillation.
Conduction system abnormalities and sudden death
Conduction system abnormalities have been described in a number of SCDs in athletes, usually due to premature aging, sclerosis of the left side of the cardiac skeleton fat, mononuclear cell infiltration and thrombosis of the sinoatrial node artery. However, for the histopathologist, the significance of these findings, together with the lack of normal controls, make a detailed autopsy study of the conduction system extremely time consuming and unsatisfactory in the investigation of most sudden death cases.
The morphologically normal heart and the ‘negative’ autopsy
Data from our pathology unit demonstrate a high prevalence of SCDs with a morphologically normal heart (23%), with no other cause of death found at autopsy. Previous studies have reported significantly lower rates, as low as 1%, with only a Spanish series reporting a figure comparable to our study (16.3%).34 The identification of a morphologically normal heart is of great importance since studies indicate that more than 50% of such SCDs are caused by malignant arrhythmias secondary to the presence of inherited ion channel disorders, usually affecting the potassium, sodium and calcium ion channels of the myocardial cells, all of which will not be seen morphologically at autopsy.
In the past decade, the emergence of the channelopathies, in particular, long QT syndrome and catecholaminergic polymorphic ventricular tachycardia (CPVT), has transformed the importance of the negative autopsy. These ion channelopathies which cannot be detected at autopsy are inherited, as are the cardiomyopathies, usually in an autosomal-dominant manner. Sudden unexpected death during exercise particularly can, in many families, bring to their attention a hitherto unsuspected cardiac condition which is inherited and may be diagnosed by screening first-degree family members. Missed diagnoses/wrong diagnoses can have catastrophic consequences for families in which other members are at risk of sudden death because they carry the defective gene for that condition.35
The incidence of cardiac events is highest in LQT1 and LQT2, but sudden death is highest in LQT3. These episodes are characteristically associated with sudden increases in sympathetic activity, such as exercise, notably swimming. CPVT is a genetic arrhythmogenic disorder characterised by stress-induced, bidirectional ventricular tachycardia that may degenerate into cardiac arrest and sudden death. Mutations in the cardiac ryanodine receptor gene have been identified in CPVT and in a subgroup of patients with ARVC, so there is an overlap with cardiomyopathies. Based on this knowledge, established treatment strategies have been introduced and affected patients are advised to avoid strenuous sports.
The pathologist is thus pivotal in providing a definitive cause of the SCD, and guiding the cardiac screening management for living relatives of the deceased individual(s). A previous study undertaken at our unit highlights the prominent role of electrical abnormalities in the normal heart in SCDs, particularly within younger patients.36 Although selection bias has certainly contributed to the high prevalence of SCDs with a morphologically normal heart, this study highlights the importance of establishing such a diagnosis, as ion channelopathies can be identified in living relatives with appropriate screening and treatment instigated to avoid further tragedies.
Summary
Sudden death is rare and rarer still in fit young individuals. Nevertheless, their deaths generate widespread public attention as they are seen as the epitome of health and fitness. The role of the histopathologist is to provide a detailed examination of the heart, in order to identify if the cause of death was due to a CAD, cardiomyopathy, valve abnormality, major vessel rupture or ion channelopathy mechanism. The histopathologist is thus vital in providing a definitive cause of the SCD, and appropriately guiding the screening management of living relatives for the deceased individual(s). For the future, it is imperative that pathologists develop standardised methods, pathological criteria for entities and provide clear reporting in all these cases. However, at the present moment it is essential that the heart of all young individuals dying unexpectedly is examined by an expert cardiac pathologist.
References
Footnotes
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.