Article Text

Abstract
Long-term high-intensity physical activity is associated with morphological changes, termed as the ‘athlete's heart’. The differentiation of physiological cardiac adaptive changes in response to high-level exercise from pathological changes consistent with an inherited cardiomyopathy is imperative. Cardiovascular magnetic resonance (CMR) imaging allows definition of abnormal processes occurring at the tissue level, including, importantly, myocardial fibrosis. It is therefore vital in accurately making this differentiation. In this review, we will review the role of CMR imaging of fibrosis, and detail CMR characterisation of myocardial fibrosis in various cardiomyopathies, and the implications of fibrosis. Additionally, we will outline advances in imaging fibrosis, in particular T1 mapping. Finally we will address the role of CMR in pre-participation screening.
- Athletics
- Cardiology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Long-term high-intensity physical activity is associated with apparent ‘physiological’ cardiac morphological changes, principally left (LV) and right ventricular (RV) enlargement, together with electrocardiographic modifications including, most commonly, resting bradycardia, repolarisation abnormalities and increased voltage suggestive of LV hypertrophy (LVH). These manifestations are classically termed as the ‘athlete's heart’ (table 1);1 and since the development of echocardiography in the 1970s and 1980s, there is now a plethora of conclusive evidence documenting the cardiac structure and function of highly trained athletes of different ages, ethnicities, genders, competing in a variety of sporting disciplines.
Alterations in morphology and function in the athletic heart1
The purpose of this review is to introduce the advanced imaging modality, cardiovascular magnetic resonance (CMR), in the evaluation of the ‘grey zone’ athlete commonly encountered when trying to differentiate physiological cardiac adaptive changes in response to high-level exercise, from pathological changes consistent with an inherited cardiomyopathy, such as dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), arrhythmogenic right ventricular cardiomyopathy (ARVC) and/or left ventricular non-compaction (LVNC). While transthoracic echocardiography is routine and easily available, it is limited in its inability to accurately define processes that are occurring at the myocardial tissue level. In contrast, CMR imaging allows definition of abnormal processes occurring at the tissue level, including myocardial oedema, fatty infiltration, and importantly, myocardial fibrosis. It is the identification of these myocardial tissue processes as well as the measurement of cardiac volumes and mass with unparalleled precision that makes CMR a unique and powerful tool for differentiating pathological from physiological LVH.
While CMR offers a number of advantages in the assessment of patients with athletic heart, this review will specifically examine the role of CMR imaging of fibrosis in athletes with left ventricular hypertrophy. First, we will address the challenges in the differentiation of the athlete's heart versus cardiomyopathy. We will then briefly overview the CMR image sequences used and the pivotal role, and limitations of late gadolinium enhancement (LGE) in imaging fibrosis. We will then outline recent advances in imaging fibrosis, in particular T1 mapping which may afford the potential to detect fibrosis that is currently missed by LGE techniques. We will briefly detail CMR characterisation of the different types of myocardial fibrosis and the implications of fibrosis. Finally, we will address future directions of CMR imaging and its role in pre-participation screening.
Challenges in the differentiation of the athlete's heart versus cardiomyopathy
A small proportion of athletes with unsuspected cardiac pathology are at increased risk of exercise-related sudden cardiac death (SCD),2 ,3 with sudden death often the first clinical manifestation of underlying heart disease.4 In healthy athletic adults (age <35 years), the incidence of sports-related sudden death ranges from 1:15 000 to 1:50 000.5 Cardiomyopathies are the commonest cause of sudden death among athletes.6 ,7 However, while studies in the USA have shown HCM to be the predominant pathology8 and ARVC in Italy,9 LVH without myocyte disarray was shown to be the cause in 31% in a comprehensive UK study.4 This study also demonstrated idiopathic myocardial fibrosis, with or without LVH, featured in 14% of cases. Whether the LVH induced by athletic training predisposes to myocardial fibrosis, which subsequently acts as a substrate for fatal ventricular dysrhythmias, remains unclear.
While a diagnosis of phenotypically expressed HCM requires disqualification from most sports to minimise the risk of sudden death,10 a mis-diagnosis of athletic heart may have fatal consequences. The differentiation between physiological LVH and HCM is thus essential, but often clinically challenging.11 The extremes of LVH seen in athletes (beyond 13 mm in men and 12 mm in women) overlap with that seen in patients with morphologically mild HCM.11 Thus, an athlete with LVH beyond 13 mm represents a grey zone between physiological adaptation and mild expression of HCM, although LVH of up to 16 mm has been shown in black athletes, which again highlights the difficulty of interpreting wall thickness measurements in making the distinction between pathology and athletic remodelling.12 Various criteria for making this distinction have been described, including the degree of LVH, patterns of LVH and left ventricular cavity size (table 2).13 However, the subtleties of differentiation between LVH and HCM may remain challenging despite extensive echocardiographic assessment, and indeed morphologically mild HCM may nonetheless be associated with significant risk of SCD. Finally, LV cavity dimensions may rarely be increased to a degree compatible with primary DCM in a minority of athletes.5 DCM is also an important cause of sudden death among young athletes.14 It is therefore essential to accurately differentiate physiological and pathological cardiac enlargement in athletes in order to prevent exercise-related SCD.
Distinguishing hypertrophic cardiomyopathy (HCM) from athletic heart
CMR versus echocardiographic imaging
Previously, in conjunction with an electrocardiogram, echocardiography was considered the standard non-invasive diagnostic test for HCM.15 ,16 However, the diffuse nature of the disease pattern in HCM limits the usefulness of echocardiography which often fails to adequately visualise the anterolateral free wall and apex.11
The distribution of hypertrophy in HCM is often asymmetrical;17 consequently, subtle segmental areas of hypertrophy may be missed on echocardiography.18 Imaging with CMR relies on the specific properties of protons in any tissue, which are determined by tissue composition. Detailed anatomical assessment is performed by imaging in multiple planes. This gives a three-dimensional representation of anatomy and therefore allows the evaluation of areas not amenable to assessment with echocardiography. In particular, CMR is vital for the assessment of apical hypertrophy and assessment of the anterolateral free wall.19 CMR is thus the reference standard imaging modality for the assessment of ventricular volumes, function, mass and tissue characterisation (eg, myocardial fibrosis).
CMR image sequences
There are three main techniques used in clinical CMR. Spin echo imaging, gradient echo imaging and flow velocity encoding. In spin echo imaging, the tissues are bright and the blood is dark (black blood). These sequences provide high-resolution images with excellent endocardial border definition of all regions of the LV, and virtually permit the reconstruction of the chamber.20 ,21 This method is thus predominantly used for anatomical assessment, and for identifying the fatty infiltration of ARVC.22 In general, images obtained with gradient echo imaging show the blood as bright and myocardium appears dark (bright blood approach). This technique is used to assess LV and RV size and function, ventricular mass, intracardiac shunts and valvular function. Steady-state free precession is related to gradient echo imaging and generates high temporal (less than 30 ms) and spatial (2 mm in-plane) resolution cine images in a single breath-hold. Finally, flow velocity encoding (also known as phase-contrast) directly measures blood flow and is used to quantify the severity of valvular regurgitation and stenosis and intracardiac shunt size.
The use of these techniques generates high spatial resolution three-dimensional images allowing precise morphological and functional assessment. CMR is particularly useful in the assessment of LV and RV mass, size and systolic function.23 The reproducibility of CMR for functional parameters is superior to that of echocardiography.24 The accuracy of CMR allows identification of subtle changes in functional parameters in patients.
LGE and imaging fibrosis
The use of contrast agents, in particular gadolinium chelates, has revolutionised the applicability of CMR in the evaluation of cardiac disease. Gadolinium-based extracellular paramagnetic contrast agents accumulate in areas of extracellular expansion and thus can be used to delineate areas of injured myocardium. Typically, areas of gadolinium accumulation relate to areas of scar expansion due to focal myocardial replacement fibrosis, such as that which occurs in both ischaemic and non-ischaemic pathologies.25 Delayed clearance of gadolinium may be quantified on T1-weighted images to diagnose areas of myocardial fibrosis.26 Gadolinium reduces hydrogen proton T1-relaxation times in proportion to its local concentration. In areas of myocardial fibrosis, there is decreased perfusion of the fibrotic tissue and thus a prolonged wash-out time for the gadolinium.27 This increased gadolinium concentration causes shortening of T1 time, appearing as bright signal intensity in the CMR image based on gradient echo sequences (ie, in T1-weighted imaging, tissues with a shorter T1-relaxation time exhibit greater signal intensity than those with longer T1-relaxation times). Late imaging (after at least 5 min post-contrast) with T1-weighted inversion recovery sequences identifies conditions associated with expansion of the extracellular space and fibrosis. In this way LGE tissue characterisation plays a crucial role in defining the pattern of fibrosis, which in turn allows identification of the underlying disease.
Limitations of LGE
While the use of LGE to identify myocardial fibrosis is sensitive, accurate quantification of the burden of fibrosis is limited.28 LGE signal differs from one study to another and therefore direct comparisons cannot be made. Second, LGE is influenced by technical parameters, including the threshold set to differentiate normal from fibrotic myocardium.29 This has resulted in variability in frequency of myocardial fibrosis in various cardiomyopathies between studies and thus LGE is unreliable for quantification of myocardial fibrosis in this setting. Finally, LGE typically images only focal macroscopic replacement fibrosis and not microscopic fibrosis. As large signal intensity differences between fibrotic and normal myocardium may not exist when the fibrosis is diffuse, LGE has limited use in the assessment of diffuse interstitial fibrosis. Furthermore, LGE techniques seem to represent a late stage of a pathological process, and there is increasing interest in the detection of early markers of an abnormal myocardial process, to which newer CMR techniques may have a role.
Recent advances in imaging myocardial fibrosis: T1 mapping
In CMR, the signal intensity is based on the relaxation after radiofrequency excitation of hydrogen protons in the static magnetic field. There are two MR relaxation parameters, T1 and T2, both measured in milliseconds, which depend on the molecular make up of tissues. These not only vary between tissues, but also within tissue depending on the presence of inflammation or fibrosis. Overall, three primary sequences are used to enhance tissue characterisation. First, T1-weighted early contrast-enhanced sequences assess myocardial hyperaemia and capillary leak.30 T2-weighted sequences assess myocardial oedema31 and T1-weighted late enhancement imaging assesses myocardial fibrosis.32 Therefore, specific CMR sequences unveil particular within-tissue changes, such as fibrosis. The use of gadolinium further enhances these changes enabling them to be more readily imaged.
LGE imaging sequences delineate fibrosis by revealing a relative difference in T1-relaxation times between areas of scar (T1 shortened by accumulation of gadolinium) and normal myocardium (T1 closer to normal as gadolinium is rapidly washed out). T1-mapping techniques work by measuring the absolute T1-relaxation time for all areas of myocardium on a pixel-by-pixel basis. As the shortening of T1-relaxation time is proportional to the local concentration of gadolinium, this can reveal subtle changes in T1 times due to expansion of the interstitial space with collagen and other fibrous tissue components. The Modified Look-Locker inversion recovery (MOLLI) sequence is a popular approach for doing this and can allow a measurement of T1 times in a single breath hold.33
As well as being influenced by the amount of scar present, the local concentration of gadolinium will be affected by the rate at which gadolinium is cleared from the body and also by the amount of extracellular fluid available in the body of the contrast to distribute into.34 With the knowledge of the patient's haematocrit, simple kinetic models exist to allow corrections to be made for these factors, generating a standardised estimate of the extracellular volume fraction, Ve, (an index of fibrosis if the extracellular space is occupied by scar tissue).35 ,36
T1-mapping has the potential to differentiate both interstitial and replacement fibrosis from normal myocardium but not one type of fibrosis from another.37 T1-mapping allows fibrosis quantification on a standardised absolute scale. It may therefore represent a more accurate means of quantifying total fibrotic burden than LGE approaches. While to date there are very few studies published using T1-mapping in the clinical setting, it is hoped T1-mapping may also reveal and allow quantitative assessment of diffuse myocardial fibrosis.
T1-mapping: future directions
Previous studies have shown that up to 50% of veteran athletes demonstrate myocardial fibrosis.38 T1-mapping techniques have the potential to identify unsuspected interstitial fibrosis in a significant proportion of athletes and veteran athletes, although their usefulness may be limited by multiple confounders in this latter age group, including hypertension and diabetes. Therefore, discriminatory techniques to accurately differentiate normal, physiologically adaptive T1 signals in athletes from potential pathology are needed. This evolution of T1-mapping over the coming years will likely mirror that of LGE over the past decade.39
Despite these significant imaging advances, challenges will arise. LVH and chamber dilatation in non-pathological hearts in high-level athletes may indeed demonstrate variable degrees of interstitial fibrosis. This may arise as a consequence of ultraendurance exercise, or possibly as a consequence of chamber remodelling, potentially therefore representing a physiological process. Nonetheless, given post-mortem data suggesting idiopathic LVH and interstitial fibrosis as the aetiology of SCD in athletes,40 one would postulate that these techniques may be useful at identifying high-level athletes who may be at risk of developing an exaggerated fibrotic response to exercise.
Pathogenesis of myocardial fibrosis
Myocardial fibrosis is a scarring process which develops in response to a cardiac insult (ischaemia, infection, inflammation or genetic abnormality). Myocardial fibrosis increases LV stiffness and reduces LV compliance, resulting in impaired systolic and diastolic function and reduced cardiac output.41 Myocardial fibrosis is characterised by fibroblast accumulation and excess deposition of extracellular matrix proteins, which leads to distorted organ architecture and function.42 Increased collagen deposition occurs as a result of an imbalance between collagen synthesis and degradation43 or an increased ratio of type I to type III collagen.44 In addition to collagen deposition, there is increased accumulation of other extracellular matrix proteins within the myocardium, including laminin and fibronectin.45 Myocardial fibrosis may be reactive or replacement. In reactive fibrosis, collagen accumulates in perivascular and interstitial tissue and is not accompanied by myocyte loss. In replacement fibrosis, there is loss of myocytes.
Mechanisms underlying myocardial fibrosis
The mechanism of development of fibrosis in athletes without an inherited cardiac disease process is unknown. Endurance exercise has been shown to induce release of cardiac troponins, which are clinically regarded as biochemical evidence of myocardial injury.26 Previously, it was postulated that raised levels of these humoral markers of cardiac myocyte damage indicated that microscopic myocardial damage occurred with exercise, and repeated bouts may have resulted in the development of myocardial fibrosis. However, whether or not the elevation of cardiac biomarkers after endurance exercise represents proof of detectable myocyte cell death remains unclear. While a general prevalence of subclinical myocardial injury of 12% in older marathon runners, independent of acute exercise, has been previously reported,46 recent CMR studies did not detect any myocardial damage by LGE in runners immediately after a marathon race.47–49 Postulated aetiologies of postexercise troponin release include enhanced membrane permeability and cytoplasmic release of myocytes,50 ventricular strain or a release of troponin from peripheral stem cells.35
Two hypotheses for the development of myocardial fibrosis have been postulated.51 The first hypothesises that myocardial injury with exercise is followed by repair and results in myocyte hypertrophy.52 Extrapolation of this hypothesis may suggest that the aetiology of fibrosis in athletes may be a result of physiological changes following the development of LVH, similar to the aetiology of fibrosis in HCM. In patients without LVH, it is unusual to find myocardial fibrosis. This suggests that myocardial fibrosis occurs after development of LVH.53 A postulated physiological basis for this finding in HCM is that increased oxygen demand from LVH results in myocyte death and replacement fibrosis54 and the LV outflow tract pressure gradient resulting from LVH causes pressure necrosis of intramural small vessel coronary arteries.55 Indeed, recent studies using stress-perfusion CMR hypothesise that these microvascular abnormalities precede and predispose to the development of myocardial fibrosis.56 Whyte et al.40 reported the presence of myocardial fibrosis and idiopathic LVH at postmortem in the heart of an athlete WHO died suddenly during a marathon race. At autopsy, the weight of the heart was 480 g (upper limit of normal of 431 g for a 75 kg man), and there was widespread replacement fibrosis particularly in the lateral and posterior ventricular walls as well as interstitial fibrosis in the inner layer of the myocardium, in the absence of myocyte disarray. The authors hypothesised that in the absence of any other cause, lifelong repetitive endurance exercise may result in fibrotic replacement of the myocardium in susceptible individuals, resulting in a pathological substrate for the development of arrhythmias, possibly reflecting an exercise-induced HCM-like fibrotic process, or indeed, HCM that was not diagnosed antemortem. This hypothesis is supported by animal work in which male rats are conditioned to run for 16 weeks. There were resultant findings of increased collagen deposition and fibrotic markers, accompanied by alteration in ventricular function and a susceptibility to arrhythmia.57
In contrast, a second hypothesis suggests that myocardial injury is followed by scarring leading to fibrotic replacement of the myocardium that is associated with an increased potential for arrhythmia generation.58 Myocardial fibrosis, in the absence of LVH or coronary atherosclerosis, may occur as the result of elevated catecholamines and coronary vasospasm leading to a cascade of ischaemia, necrosis and fibrosis.59 ,60
Myocardial fibrosis (both interstitial and replacement) has been shown to be a potential mechanistic substrate and marker of disease state.61 ,62 In postmortem series, replacement fibrosis is detected in nearly 60% of patients with HCM who died suddenly, with the collagen network found to be eight times greater in patients with HCM than in controls.63 ,64 The presence of fibrosis contributes to the disruption of the electrical synchrony between myocytes and therefore increases arrhythmic potential.65 ,66 It also promotes increased myocardial stiffness with LV diastolic dysfunction.67 This is followed by adverse remodelling leading to cavity dilatation and eventually systolic dysfunction, which is detectable in 85% of patients with end-stage dilated HCM.68 Therefore, its accurate and early identification is of the utmost clinical importance.
Implications of fibrosis
As outlined previously, athletes typically develop various degrees of LVH, often eccentric and associated with increases in LV end-diastolic and end-systolic dimensions. Thus, three cardiomyopathies that are of clinical importance in regard to the evaluation of the ‘grey zone’ athlete include HCM, DCM and ARVC. It should be noted that some athletes with obvious phenotypic HCM expression can achieve high-level physical performance. Thus, their athletic prowess should not be used as a discriminator between physiological and pathological remodelling. CMR is therefore essential to accurately identify the pattern of fibrosis seen in inherited cardiomyopathies, such as HCM, DCM, ARVC and LVNC (table 3).
Added value of CMR in the diagnosis and differentiation of cardiomyopathies
Hypertrophic cardiomyopathy
HCM is a genetic disorder characterised by the development of cardiac muscle fibre hypertrophy, disarray, dysplasia of intramural coronary arterioles and myocardial fibrosis. HCM may be differentiated from LVH associated with athlete's heart based on the maximum end-diastolic wall thickness-to-volume ratio (maximal end-diastolic wall thickness/indexed LV end-diastolic volume). An end-diastolic wall thickness to volume ratio of <0.15 mm/m2/ml was shown to have a 99% specificity in differentiating athlete's heart from HCM.69
Myocardial fibrosis or scar detected by CMR occurs in up to 33–86% of patients with HCM.70 Fibrosis in HCM is patchy and occurs predominantly within hypertrophied segments. Typically this fibrosis is seen at the junction of the right ventricle and interventricular septum.54 The prognostic significance of the presence of fibrosis, as demonstrated by LGE, to adverse outcome is high and has been associated with sudden cardiac death, systolic dysfunction and non-sustained ventricular tachycardia.71 ,72 The extent of fibrosis has been shown to be a predictor of arrhythmic events,73 ,74 and correlated with risk factors for SCD and the likelihood of inducible VT75 (figure 1).

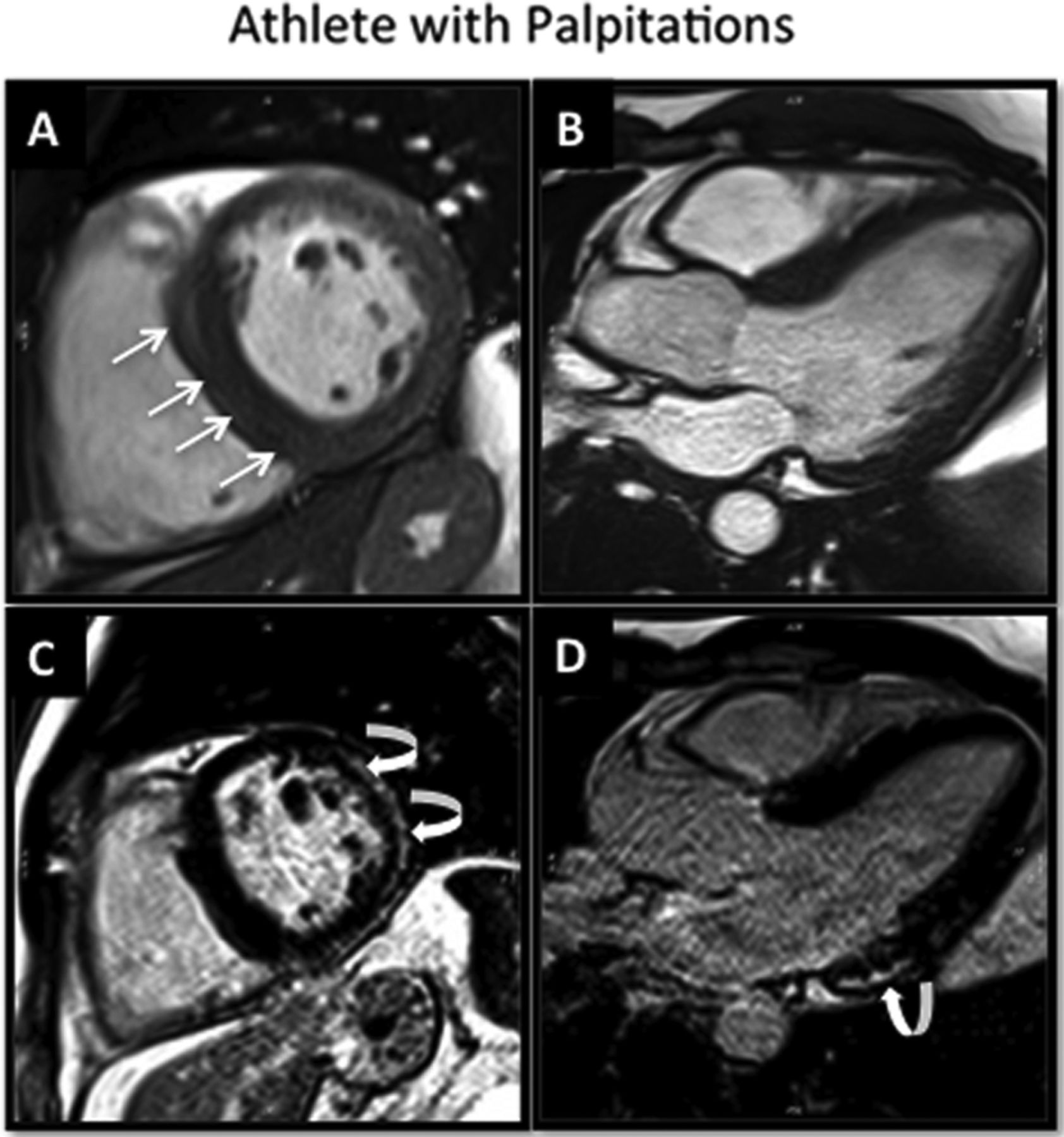
High level athlete with asymmetrical left ventricular hypertrophy (basal septum). Normal ECG. Recent palpitations and frequent premature ventricular contractions on holter. Echocardiogram suggestive of hypertrophic cardiomyopathy (HCM). Steady-state free precession cine images (A, B) demonstrate mild septal hypertrophy but prominent right ventricular septomarginal trabeculation (straight arrows) falsely giving the impression of HCM. The late gadolinium enhanced images (C,D) demonstrate regions of epicardial fibrosis in the inferior and lateral walls (curved arrow) consistent with a diagnosis of myocarditis in an athlete's heart.
Should the differentiation between HCM and athlete's heart still remain unclear following CMR, the role of deconditioning emerges. A repeat CMR following a three-month period of deconditioning, with precise wall thickness assessment at baseline and following deconditioning, should show regression of LVH in athlete's heart and not in HCM, and thus allow accurate differentiation.
Dilated cardiomyopathy
DCM is characterised by an increase in end-diastolic volume and reduced systolic function of predominately the ventricle. At a pathological level, there is replacement of cardiomyocytes by fibrotic tissue. Indeed, autopsy studies have shown that interstitial fibrosis is present in at least 57% of cases of non-ischaemic DCM and that up to 20% of the LV myocardial mass may be scar in these cases.54 CMR is an important tool for defining the aetiology of DCM. Ischaemic DCM shows subendocardial extending to transmural LGE generally restricted to the perfusion territory of one coronary artery. McCrohon et al.76 first demonstrated that LGE in non-ischaemic DCM has a patchy, mid-wall distribution in 28% of cases but in 13% has a subendocardial pattern indistinguishable from ischaemic cardiomyopathy. In patients with DCM, mid-wall LGE is a significant predictor of cardiac death, appropriate ICD discharge and hospitalisation for acute decompensated heart failure.77 In athletes, often, the clear distinction of the overlap between dilating chambers and hypertrophy is difficult to make. Most athletes typically have training regimes, which combine endurance cardiovascular exercises with resistance isotonic weight training. However, the presence of patchy focal intramyocardial fibrosis, as well as mid-wall fibrosis has not been demonstrated in the true remodelled athletic heart. Hence CMR, utilising LGE, may be useful to differentiate pathological hypertrophy or chamber dilatation from hypertrophy and chamber dilatation due to athlete's heart (figure 4).

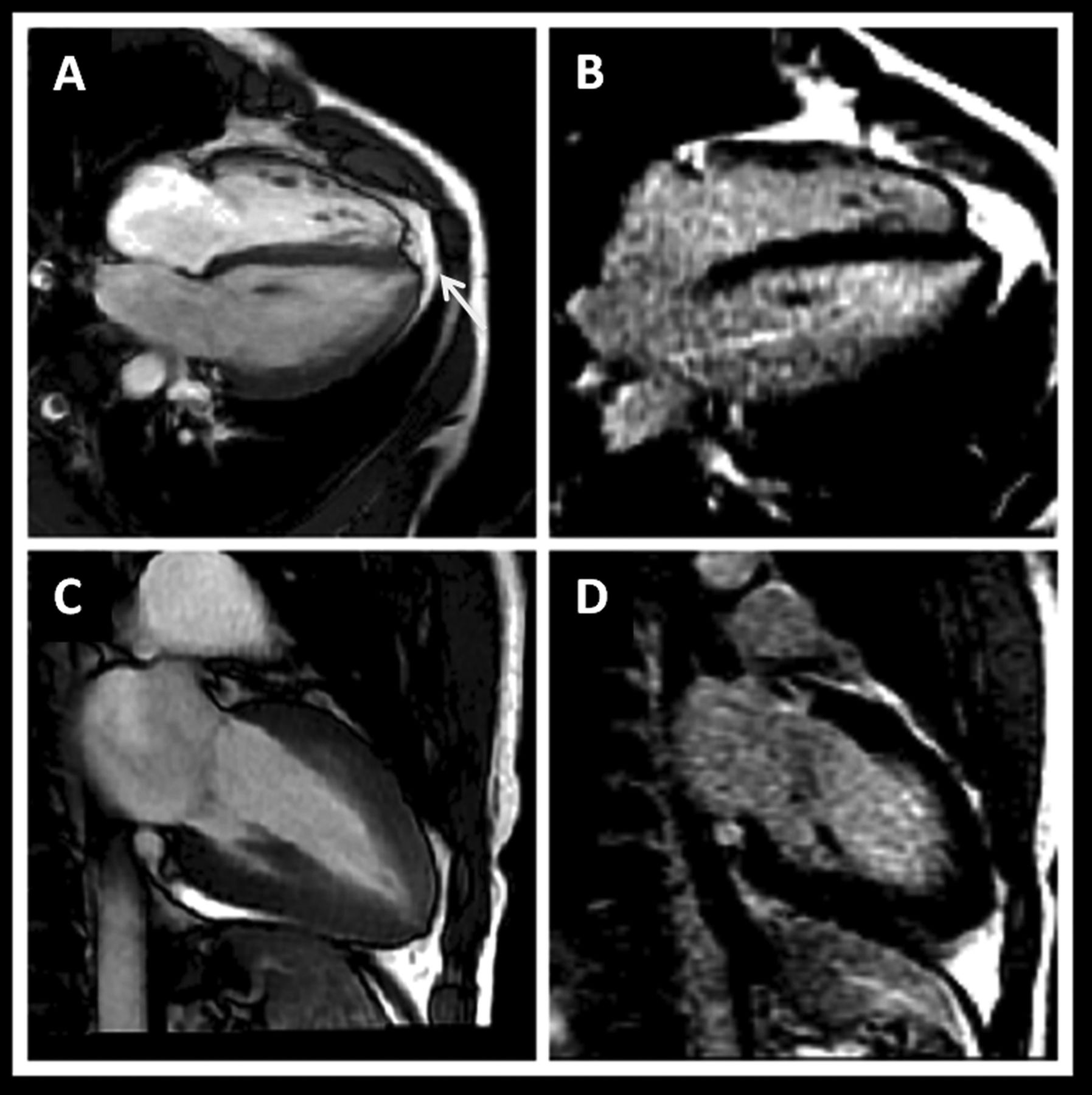
Cardiovascular magnetic resonance (CMR) in the athlete with an abnormal ECG. High-level boxer with grossly abnormal ECG (deep T-wave inversion throughout all leads), normal echocardiogam and asymptomatic. Steady-state free precession cine CMR (A,C) demonstrates subtle apical hypertrophic cardiomyopathy (white arrow) without evidence of myocardial fibrosis on late gadolinium enhanced images (B,D).
Abnormal RV
The most commonly encountered arrhythmias are those originating from the RV.78 Although in most cases RV arrhythmias arise from a structurally normal heart and carry a benign prognosis, they may also be the manifestation of an underlying cardiomyopathy such as ARVC.79 RV outflow tract-ventricular tachycardia and chronic RV remodelling have been described in amateur athletes following marathon running.80 However, RV arrhythmias and RV impairment are most commonly seen in highly trained, ultraendurance athletes,81 and unlike amateur athletes, these may persist despite detraining.82 A recent study hypothesises that, in veteran endurance athletes, the ARVC phenotype by Task Force criteria, may be acquired through intensive and sustained endurance exercise and may not be solely attributable to a genetic predisposition.83 Given the significant consequences of undiagnosed ARVC, the differentiation between cardiomyopathy-related and idiopathic RV arrhythmia is crucial. CMR has become the gold standard imaging modality for assessing these patients as it allows accurate analysis of morphological and functional evaluation, as well as definitive myocardial tissue characterisation.84 However, CMR evaluation of athletes for ARVC is complicated by overlapping features such as RV volume increase. Recent revised Task Force Criteria have been published which detail that the distinction hinges on the fact that athletes show proportionate changes in LV and RV volumes while patients with ARVC demonstrate disproportionate changes.85 Given that CMR is the gold standard for LV and RV volume analysis, this distinction is thus most accurately addressed by CMR (figure 5).

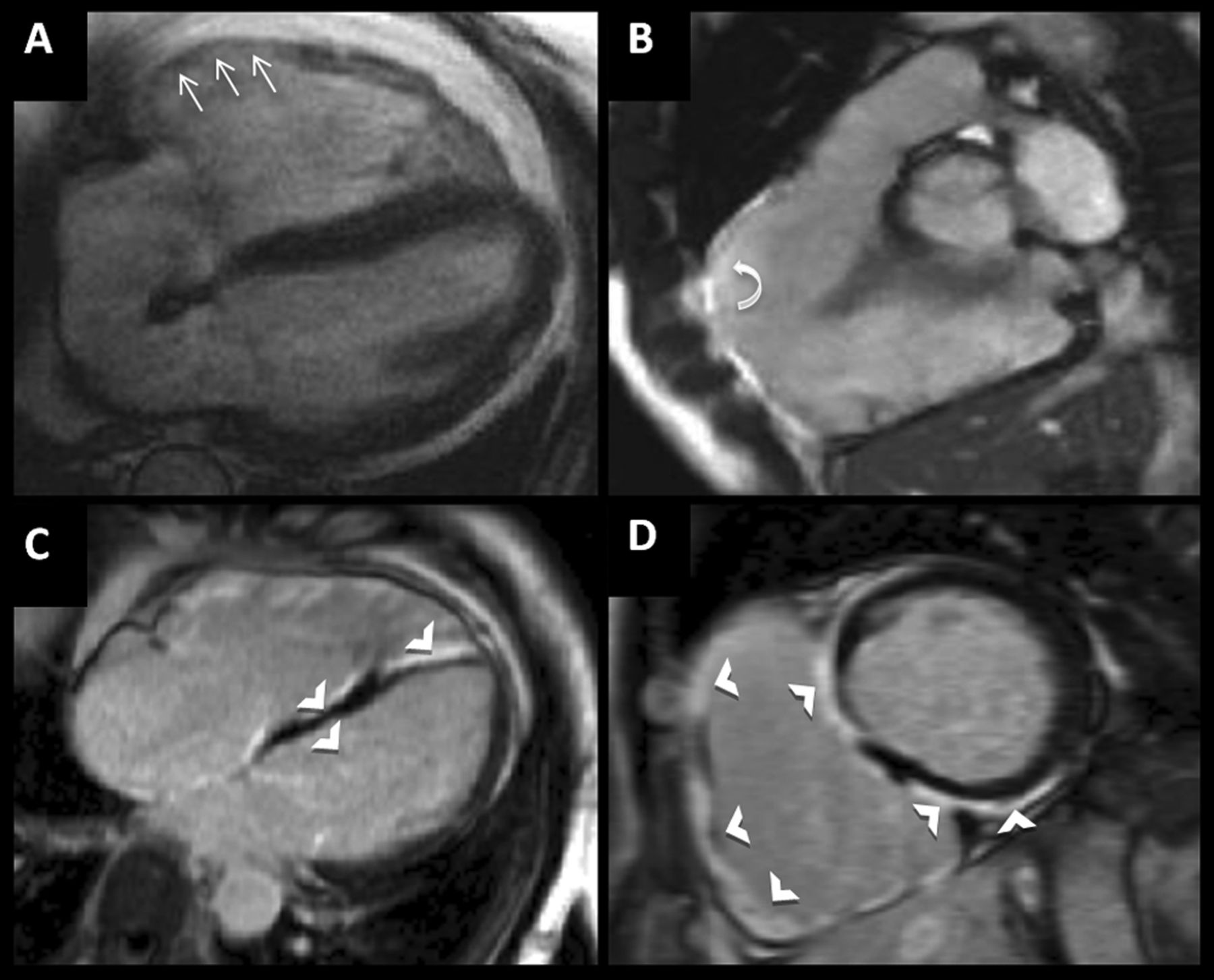
High-level athlete with abnormal ECG (deep T-wave inversion in lateral leads) and asymptomatic. No family hx. Echocardiogram suggests concentric left ventricular hypertrophy (LVH). Cardiovascular magnetic resonance performed to differentiate athletic remodelling from cardiomyopathy. Steady-state free precession cine images (top row, A–D) demonstrating mild concentric LVH with the short axis cine slice (D) showing asymmetrical thickening of the basal septum. Late gadolinium-enhanced images (E, I) demonstrating patchy mid-wall enhancement consistent with myocardial fibrosis (white arrows) diagnostic of hypertrophic cardiomyopathy.
Left ventricular non-compaction
LVNC is characterised by the presence of an extensive non-compacted myocardial layer lining the cavity of the LV and potentially leads to cardiac failure, thromboembolism and malignant arrhythmias.85 Pathological studies have previously demonstrated areas of myocardial fibrosis in patients with isolated LVNC,86 supported by recent CMR LGE studies.87 ,88 Indeed, one recent study has shown fibrosis by LGE in 55% of isolated LVNC patients.89 This study also demonstrated a significant burden of fibrosis with fibrosis typically involving 5% of the overall LV myocardium. Fibrosis was present in similar prevalence in both compacted and non-compacted segments, supporting the hypothesis that LVNC may indeed be a marker of an underlying diffuse cardiomyopathy, involving both normal and non-compacted myocardium,90 ,91 rather than a disease entity in and of itself (figure 6). However, hypertrabeculation may also be observed in the absence of LVNC. Ethnicity is an important determinant of hypertrabeculation.92 Athletes of black ethnicity have significantly more pronounced ventricular hypertrabeculation, resembling LVNC. As this hypertrabeculation is likely physiological, it is important to accurately differentiate physiological hypertrabeculation due to cardiac adaption, from LVCC. CMR LGE is essential for this distinction (figure 7).

Dilated cardiomyopathy. Cine image (A) showing dilated left ventricular with wall thinning and mild increase in lateral wall trabeculation. Late gadolinium images (B) show typical mid-wall enhancement (white arrows), confirmed macroscopically (C) as myocardial fibrosis.
Myocarditis
Myocarditis may result in death from ventricular arrhythmias. Differentiating LV dilatation from myocarditis from that due to athletic training is challenging and therefore CMR assessment of myocardial fibrosis is crucial in making the differentiation. The fibrosis pattern seen myocarditis is often patchy does not necessarily involve the subendocardium.93 ,94 The epicardium of the inferior lateral wall has previously been shown to be the most commonly affected area.95 ,96 LGE changes may be seen early in the disease and regress with resolution of symptoms.97 ,98
Finally, it should, of course, be noted that additional causes of death in athletes would include anomalous origin of the coronary arteries, QT-interval prolongation syndromes and mitral valve prolapse, aortic valve stenosis, among others (figure 5, 6, 7).

A patient with arrhythmogenic right ventricular cardiomyopathy. Cine images (A,B) demonstrate a dilated right ventricle (RV) with focal wall thinning of the basal RV free wall (thin arrows) and a localised aneurysm best seen in the RV outflow tract (curved arrows). Late gadolinium-enhanced images (C,D) demonstrate extensive myocardial fibrosis in the RV septum and RV free wall extending also to involve the left ventricular inferior wall (possible arrhythmogenic left ventricular cardiomyopathy overlap) (thick arrows).

In this patient with left ventricular non-compaction, there is marked left ventricular apical and lateral wall trabeculations. Additionally, fibrosis is seen to be present in similar prevalence in both compacted and non-compacted segments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Challenges in physiological from pathological left ventricular trabeculations (white arrows) and non compaction (A–D). Images A and B from an athlete with increased trabeculations consistent with physiological remodelling. Images C and D from a patient with left ventricular non-compaction (LVNC) with more marked regions of non-compact myocardium fulfilling criteria for LVNC.
Future directions for pre-participation screening
Imaging fibrosis in junior athletes
In the USA, HCM accounts for up to one-third of all deaths in young athletes. Therefore, accurate differentiation between physiological LVH and HCM is imperative in this population. ECG screening of athletes is emerging in some countries.93 ECG screening of athletes has however been controversial. While Corrado et al95 demonstrated that the annual incidence of SCD among athletes reduced significantly from 3.9 per 100 000 person-years to 0.4 per 100 000 person years between 1979 (preimplementation) and 2004 (postimplementation), a recent study demonstrated that the incidence of SCD did not decline following the introduction ECG screening.97 Given these conflicting findings, there may be a role for structural assessment to detect the most common causes of SCD. Transthoracic echocardiography is the primary imaging modality used to assess for HCM, DCM, ARVC and myocarditis. However, if the images obtained yield insufficient information to exclude cardiac pathologies, additional alternative modalities, namely CMR, may be considered in selected at-risk individuals. Indeed, while the integration of CMR into the screening pathway would provide a comprehensive evaluation of young athletes found to have abnormalities on ECG, its integration into routine pre-participation is unlikely given the significant cost, and logistical limitations to such a strategy.
Veteran athletes
The consequence of long-term prolonged endurance exercise in veteran athletes is incompletely understood. It has been shown that, in the absence of other causes, endurance exercise may result in myocardial fibrosis, which then acts as a substrate for arrhythmias.83 ,98 This hypothesis is supported by a previously described case of sudden death in a veteran athlete during marathon running.40 Given that up to 50% of veteran athletes may have unsuspected myocardial fibrosis,83 ,99 and thus carry the consequent risks, there may be a role for pre-participation screening and subsequent risk stratification of veteran athletes. Of course, further work and larger scale clinical trials are required to identify the exercise threshold so that at-risk individuals may be identified and an appropriate imaging strategy may be designed, and importantly, limited to a defined population at risk.
Conclusion
If the tragedy of SCD in athletes is to be prevented, we must better understand the mechanisms of these events and accurately identify those at risk. In this regard, pre-participation screening, of both young and old athletes, is of utmost importance. The current recommendations of pre-participation evaluation with a 12-lead ECG aim to identify the majority of potentially life-threatening cardiovascular conditions. In those athletes who have an abnormality on ECG, CMR will have an emerging and growing role as a specific and efficient screening tool for detection of disease processes which carry a risk of SCD, and importantly will detect unsuspected myocardial fibrosis in athletes, and thus may prove crucial to prevent SCD.
References
Footnotes
-
Contributors All authors contributed throughout the concept design, drafting and editing of this manuscript.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.