


Abstract
Acquisition of androgen independence by prostate cancer is the key problem of prostate cancer progression. Vasoactive intestinal peptide (VIP), a neuropeptide, may act as a survival factor for prostate cancer cells under androgen deprivation. However, the molecular mechanisms by which VIP promotes the androgen-independent growth of androgen-sensitive prostate cancer cells have not been addressed. We therefore investigated the biological effect and signal pathway of VIP in LNCaP cells, a prostate cancer cell line that requires androgens for growth. We showed that low nanomolar concentrations of VIP, acting through Gs-protein-coupled VIP receptors, can induce LNCaP cell growth in the absence of androgen. Blockade of androgen-receptor (AR) in these cells by AR antagonist bicalutamide or by anti-AR small interfering RNA, inhibited the proliferative effect of VIP. In addition, VIP stimulated androgen-independent activation of AR with an EC50 of 3.0 ± 0.8 nM. We then investigated VIP-stimulated signaling events that may interact with the AR pathway in prostate cancer cells. VIP regulation of AR activation, mediated by VIP receptors, was protein kinase A (PKA)-dependent, and extracellular signal-regulated kinase 1/2 (ERK1/2) activation contributes to VIP-mediated AR activation. Furthermore, PKA-dependent Rap1 activation is required for both ERK1/2 activation and androgen-independent AR activation in LNCaP cells upon VIP stimulation. Finally, we showed that VIP-induced AR activation was also present in prostate cancer CWR22Rv1 and PC3 cells transfected with the wild-type AR. Altogether, we demonstrate that VIP acting through its Gs-protein-coupled receptors can cause androgen-independent transactivation of AR through a PKA/Rap1/ERK1/2 pathway, thus promoting androgen-independent proliferation of androgen-sensitive prostate cancer cells.
Prostate cancer is the most commonly diagnosed noncutaneous cancer in American men and the second leading cause (after lung) of cancer mortality. In its early stages, prostate cancer cell growth is dependent on androgens and their androgen receptor (AR), a member of the nuclear receptor superfamily (Heinlein and Chang, 2004). Thus, androgen-deprivation therapies that block AR activation cause repression of prostate tumors. Unfortunately, the majority of prostate cancers eventually transit from being androgen-dependent to androgen-independent (hormone refractory), making androgen-deprivation therapies ineffective (Feldman and Feldman, 2001).
The precise mechanisms underlying this conversion of prostate cancer to a state of androgen-independence remain an active area of research. This cannot be ascribed simply to the loss of AR expression because most hormone-refractory prostate tumors still express functional AR and require AR for proliferation (Zegarra-Moro et al., 2002). Therefore, some hormone-refractory prostate cancers are believed to be caused by androgen-independent activation of AR (Heinlein and Chang, 2004). Early research focused on investigations of molecular alterations such as AR overexpression (20% of hormone-refractory prostate cancers) or mutations in AR that could allow it to be activated by other ligands or even antiandrogens (Feldman and Feldman, 2001). However, the frequency of AR mutations in localized prostate cancer is low (<10%) even after developing hormone resistance, suggesting that AR mutations may not be the primary cause of tumor progression (Taplin et al., 2003). Alternatively, AR can be indirectly activated by factors other than androgens. These factors often activate cell-surface receptors such as G-protein-coupled receptors (GPCRs) (Daaka, 2004).
Basic and clinical research results demonstrate that GPCR systems may be excessively activated in subsets of malignant prostate cancers caused by abnormally elevated ligands for GPCRs and/or overexpression of receptors (Daaka, 2004). Kasbohm et al. (2005) demonstrated that Gs-coupled β-adrenergic receptor stimulation can transactivate AR in prostate cancer cells. It is interesting that advanced prostate cancers often have increased numbers of neuroendocrine cells that secrete neuropeptides, whereas the expression of neutral endopeptidase (NEP), a cell surface peptidase that degrades neuropeptide growth factors, is decreased in prostate cancer cells (Papandreou et al., 1998). These neuropeptides, acting through their GPCRs, result in androgen-independent AR activation, thus promoting the progression of prostate cancer cells (Shah et al., 1994; Lee et al., 2001). In addition, we reported recently that regulator of G-protein signaling 2, an inhibitor of GPCR function, attenuates androgen-independent AR signaling in prostate cancer cells, thereby inhibiting androgen-independent prostate cancer cell growth (Cao et al., 2006). In fact, inhibition of the excessively activated GPCRs has therapeutic benefits for patients with malignant prostate cancer (Nelson, 2003). Thus, identifying the ligands of GPCRs that can transactivate AR in prostate cancer becomes essential for effective treatment of advanced prostate cancers.
Vasoactive intestinal peptide (VIP) is a prominent neuropeptide with a relatively rich expression in the human prostate in association with the dense innervation by hypogastric and pelvic nerves that modulates prostate function (Polak and Bloom, 1984). VIP binds to two Gs-protein-coupled receptors (VPAC1-R and VPAC2-R), which have also been identified in normal human prostate cells and tissues and are overexpressed in malignant prostate tissues (Collado et al., 2005). VIP stimulates prostatic secretion of prostatespecific antigen (PSA), a marker for the early detection of prostate cancer, and increases the proliferation of prostatic epithelial cells in culture (Rekasi et al., 2000), suggesting that VIP is also involved in prostate carcinogenesis. In addition, VIP is known to potentiate the invasive capacity of androgen-sensitive prostate cancer cells (Rekasi et al., 2000) and acts as a survival factor for prostate cancers under androgen-deprived conditions (Sastry et al., 2006). However, the ability of VIP to transactivate AR and promote androgen-independent growth of androgen-sensitive prostate cancer cells is undefined.
The LNCaP cell line is the most widely studied cell line in prostate cancer research because it behaves much like early prostate cancer by being androgen-sensitive and having the ability to be transformed into androgen-independent cell lines both in vivo and in vitro. In the present study, using LNCaP cells as a model, we demonstrate that low nanomolar concentrations of VIP acting through VPAC-Rs can transactivate AR, thus promoting AR-dependent proliferation of androgen-sensitive prostate cancer cells in the absence of androgens. We also show that the transactivation of AR by VIP is dependent on ERK1/2 activation, and our data support a model having PKA-dependent Rap1 activation as a critical intermediate step in the signal transduction pathway between VPAC-R stimulation and ERK1/2 activation.
Materials and Methods
Cell Culture. LNCaP cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in RPMI 1640 medium supplemented with 5% fetal bovine serum (FBS) in a humidified incubator at 37°C with 5% CO2. Androgen-depleted cells were prepared by incubation in phenol red-free RPMI 1640 supplemented with 5% charcoal-stripped fetal bovine serum (CSS) for 48 h before initiation of the experiment. For experiments determining the role of protein kinases, the protein kinase inhibitors were added to the cell medium 30 min before VIP or DHT treatment.
Reagents. VIP and VIP receptor antagonist [d-P-Cl-Phe(6)-Leu(17)]-VIP were purchased from American Peptide (Sunnyvale, CA). Bicalutamide (Casodex) was a gift from Astra-Zeneca Pharmaceuticals (Cheshire, UK). The mouse IgG, mouse anti-AR antibody, mouse anti-VPAC2-R, rabbit anti-PSA antibody, polyclonal anti-β-actin antibody, anti-HA antibody and an enhanced chemiluminescence reagent kit were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-Myc antibody was from Upstate Biotechnology (Lake Placid, NY). Anti-phosphorylated p44/42 MAP kinase (active ERK1 and 2) antibody and anti-ERK1/2 total protein antibody were obtained from Cell Signaling Technology (Danvers, MA). The MAPK kinase (MEK) inhibitor U0126, the PKA inhibitor H89, and the myristoylated protein kinase inhibitor amide 14-22 (PKI), the PKC inhibitor GF109203X, the PI3K-Akt inhibitor Ly294002, the Src kinase inhibitor PP2, the epidermal growth factor (EGF), and the anti-green fluorescent protein (GFP) antibody were purchased from Calbiochem (San Diego, CA). CellTiter 96 Aqueous One solution and a PepTag Nonradioactive PKA assay kit were obtained from Promega (Madison, WI). Forskolin, phosphoramidon, 8-CPT-cAMP, 8-CPT-2Me-cAMP, and poly(deoxyinosinic-deoxycytidylic acid) were purchased from Sigma-Aldrich (St Louis, MO). Nuclear and Cytoplasmic Extraction Reagent was purchased from Pierce Chemical Company (Rockford, IL). The IRDye-labeled double-stranded AR consensus binding motif was from LI-COR Biosciences (Lincoln, NE). The androgen DHT was a gift from Dr. Zhaoyi Wang (Cancer Center, Creighton University, Omaha, NE). All reagents without a source listed were purchased either from Sigma-Aldrich or Fisher Scientific (Pittsburgh, PA).
Plasmids. ARE3-tk-LUC, a plasmid with three copies of the androgen response element (ARE) upstream from a luciferase reporter gene, was a gift from Dr. Li-Hua Wang (National Cancer Institute, Frederick Cancer Research and Development Center, Bethesda, MD). The Renilla reniformis luciferase expression plasmid, pRL-tk, was provided by Dr. Zhaoyi Wang (Cancer Center, Creighton University). pADneo2 C6-BGL, a gift from Dr. A. Himmler (Boehringer Ingelheim Research and Development, Vienna, Austria), contains six copies of the cAMP response element (CRE) upstream from the luciferase reporter gene. Plasmid pFA2-Elk1 and the reporter plasmid pFR-LUC were from Stratagene (La Jolla, CA). An empty expression vector, pcDNA3.1, was purchased from Invitrogen (Carlsbad, CA). Myc-ERK2, dominant-negative MEK1 mutant MEK1-K97M, dominant-negative ERK2 mutant ERK2-K52R, a wild-type ERK2-MEK1, and constitutively active ERK2-MEK1-LA were kind gifts from Dr. M. H. Cobb (University of Texas Southwestern Medical Center, Dallas, TX). GFP-tagged Rap1GAP plasmid was a gift from Dr. P.J. Casey (Duke University Medical Center, Durham, NC). HA-Rap1 and VPAC2-R plasmids were obtained from the University of Missouri-Rolla cDNA Resource Center (Rolla, MO). Dominant-negative Flag-Rap1-AGE mutant was a gift from Dr. H. Kitayama (Kyoto University Graduate School of Medicine, Kyoto, Japan), and the plasmid encoding wild-type AR was provided by Dr. Zafar Nawaz (Braman Family Breast Cancer Institute, Miami, FL).
Transfection and Luciferase Reporter Assays. Cells were transiently transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The transfection efficiency was routinely ∼30%, as determined by fluorescence of cells infected with the plasmid encoding GFP. For AR activity assay, 0.15 μg of ARE3-tk-LUC construct was cotransfected with pcDNA3.1 empty vector or expression vectors as indicated into LNCaP cells in 24-well plates. The total amount of plasmid DNA used was normalized to the same amount by the addition of empty plasmid. R. reniformis luciferase expression plasmid, pRL-tk (100 ng), was used as an internal control for transfection efficiency. Luciferase activities in cell lysates were measured with a Sirius luminometer (Berthold, Germany) using the dual luciferase assay system (Promega) and were normalized by the R. reniformis activities of the samples. The results are presented as -fold induction, which is the relative luciferase activity (RLU; ratio of reporter luciferases/R. reniformis luciferases) of the treated cells over that of the control cells.
For cAMP assay, a reporter plasmid containing the luciferase gene under the transcriptional control of multiple units of CRE (pADneo2-C6-BGL) was used as an index of the cAMP-dependent signaling pathway. LNCaP cells were grown to 90% confluence in 24-well plates. pADneo2-C6-BGL (0.15 μg) was routinely cotransfected with pcDNA3.1 empty vector into LNCaP cells. Eighteen hours after transfection, cells were treated with VIP at the indicated concentrations for 6 h, and cell lysates were then assayed for luciferase activity.
To monitor ERK activities, we used the Stratagene PathDetect reporter system, which contains an expression plasmid (pFA-Elk1) encoding the chimeric protein GAL4-Elk1, a phosphorylation target of activated ERKs, and a second luciferase reporter plasmid (pGAL4-LUC). Upon activation, the ERK-phosphorylated GAL4-Elk1 fusion protein will induce the expression of the luciferase reporter gene (Cao et al., 2006).
To examine the effect of Rap1GAP on VIP-stimulated ERK1/2 activity (Fig. 6A), nucleofection of LNCaP cells with Rap1GAP plasmids was conducted with a Cell Line Nucleofector Kit V (Amaxa Biosystems, Gaithersburg, MD) according to the manufacturer's instructions. The transfection efficiency was approximately 70%. Lysates from LNCaP cells treated with or without VIP were subjected to Western blot analysis of the phosphorylation status of ERK1/2.
siRNA Transfection. LNCaP cells were grown in phenol redfree RPMI 1640 containing 5% CSS for 24 h. Cells were either mock-transfected or transfected with a synthesized AR-specific siRNA SMARTpool (Upstate, Charlottesville, VA). A scrambled negative siRNA duplex (Ambion, Austin, TX) was used as control. All siRNA duplexes were transfected at a final concentration of 100 nM using Lipofectamine 2000. Suppression of AR protein expression in LNCaP cells by siRNA SMARTpool was examined by Western blot analysis.
Protein Extraction, Immunoprecipitation, Electrophoresis, and Western Blot Analysis. Protein was routinely extracted from exponentially growing cells using 1× radioimmunoprecipitation assay complete lysis buffer (Santa Cruz Biotechnology). For detection of Myc-ERK2, equal amounts of cell lysates per condition were immunoprecipitated with agarose-conjugated anti-Myc antibody for 5 h at 4°C in lysis buffer A containing 50 mM HEPES, pH 7.5, 10% glycerol, 1% Nonidet P-40, 150 mM NaCl, 2 mM MgCl2, protease inhibitors (10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride), and phosphatase inhibitors (1 mM NaF and 200 μM sodium orthovanadate). Protein samples and prestained protein standards were loaded on 10% SDS polyacrylamide gels, electrophoresed, and transferred to a PVDF membrane (pore size, 0.45 mm; Immobilon, Millipore, Billerica, MA). Immunoblots were probed with antibodies in a 10% blocking solution and developed according to instructions in the enhanced chemiluminescence Western blotting detection kit. A monoclonal antibody specific against the phosphorylated ERK1/2 was used to determine the activation of ERK1/2, whereas total ERK1/2 protein was blotted as a control. Cell lysates were also blotted by anti-PSA antibody to determine the effect of VIP or DHT treatment on endogenous PSA expression in LNCaP cells. Anti-β-actin antibody was used to detect β-actin in lysates as loading controls. The results illustrated are from representative experiments repeated at least three times.
Nonradioactive in Vitro Assay for PKA Activity. LNCaP cells grown in six-well plates were pretreated without or with H89 (1-30 μM) for 30 min. Cells were stimulated with VIP (20 nM) for 10 min and then were lysed in 0.4 ml/well lysis buffer A. The lysates were cleared from insoluble material by centrifugation at 20,000g for 10 min and were subjected to a kinase reaction with the fluorescencelabeled PKA substrate kemptide (Promega) according to the manufacturer's protocol. The reaction was stopped by boiling the samples for 10 min. The phosphorylated kemptide was separated from nonphosphorylated kemptide by 0.8% agarose electrophoresis and visualized under UV light.
Rap1 Activation Assays. Active Rap1 was assayed in an in vitro pull-down assay using Rap1 Activation Assay Kit (Upstate). In brief, LNCaP cell lysates were centrifuged at 500g at 4°C for 10 min to remove nuclei and cell debris. RalGDS-RBD (10 μg) coupled to agarose beads was added to the supernatant and the mixture incubated at 4°C for 45 min with gentle agitation. Beads were washed twice, and samples were subjected to 10% SDS-polyacrylamide gel electrophoresis followed by immunoblot with the Rap1-specific antibody.
Immunofluorescence. LNCaP cells grown on glass coverslips were starved in phenol red-free RPMI 1640 for 24 h and subsequently treated with VIP or DHT for 2 h. The cells were then fixed in cold methanol for 10 min and reconstituted in 4% normal serum in 0.1% Tween 20/PBS for 20 min. Primary monoclonal AR antibody (Santa Cruz Biotechnology) was used at a dilution of 1:100 and incubated for 2 h at 37°C followed by 3 × 10-min PBS washes. Anti-mouse secondary antibodies conjugated to fluorescein isothiocyanate were used. Microscopy images were obtained using a Nikon TS100 fluorescence microscope and Optronics digital camera (Nikon, Tokyo, Japan). For each experimental condition, fluorescence distribution patterns similar to the image shown were observed in the majority (>80%) of cells inspected.
Electrophoretic Mobility Shift Assay. LNCaP cells grown in phenol red-free RPMI 1640 medium were treated with 20 nM VIP or DHT for 2 h. Before harvest, cells were washed twice in cold PBS. Homogenates were separated into cytoplasmic and nuclear fractions using the Nuclear and Cytoplasmic Extraction Reagent (Pierce). Fractions were assayed for total protein using the Pierce BCA protein assay.
Electrophoretic mobility shift assay (EMSA) was done using the Odyssey Infrared Imaging System with AR IRDye-labeled oligonucleotide, both from LI-COR Biosciences. A quantity of 5.0 μg of nuclear protein extract was incubated with 1 μl of the IRDye-labeled double-stranded AR consensus binding motif (25 fmol) for 30 min at room temperature in a 10-μl solution containing 10 mM Tris, pH 7.5, 50 mM NaCl, 4% glycerol, 0.1% Tween 20, 2 mM dithiothreitol, 1 mM EDTA, and 100 μg/ml poly(deoxyinosinic-deoxycytidylic acid). The protein-DNA complexes were resolved on a 3% nondenaturing polyacrylamide gel containing 2.5% glycerol in 0.5× Tris-Gly-EDTA buffer at 300 V for 20 min. Gel imaging was carried out using the Odyssey at a wavelength of 700 nm. For the supershift experiment, 5.0 μg of cell nuclear extract protein were incubated with 400 ng of the monoclonal AR antibody or nonimmunized mouse IgG (Santa Cruz Biotechnology) for 2 h on ice before incubation with the IRDye-labeled probe.
Cells Growth Assays. LNCaP cells were seeded into 96-well plates at a density of 5 × 103 cells/well in RPMI 1640 phenol red-free medium supplemented with 5% FBS or with 5% CSS. Cells were treated with the indicated concentration of VIP or DHT for 48 h in the presence or absence of the VPAC-R antagonist [d-P-Cl-Phe(6)-Leu(17)]-VIP (2 μM) or the AR antagonist bicalutamide (20 μM). Cell proliferation was measured using the CellTiter96 Aqueous One solution assay according to the manufacturer's recommendations. In parallel experiments, cells were cultured in 5% FBS or 5% CSS without or with 20 nM VIP or DHT for 4 days and counted every 2 days using a Coulter Counter ZM (Coulter Electronics, Miami, FL). Results represent an average of at least five independent experiments performed in triplicate.
To examine the effect of silencing AR on LNCaP cell growth, cells transfected with AR-specific siRNA or scrambled siRNA were grown in phenol red-free RPMI 1640 containing 5% CSS for 48 h. These cells then were treated without or with 20 nM VIP or DHT for an additional 48 h, and total cell numbers in each well were counted. Cell growth is expressed as a percentage of the cells contained in control wells (untreated mock-transfected cells).
Statistical Analysis. Results are expressed as the mean ± S.E. of at least three determinations and statistical comparisons are based on the Student's t test. A p value of <0.05 was considered to be significant.

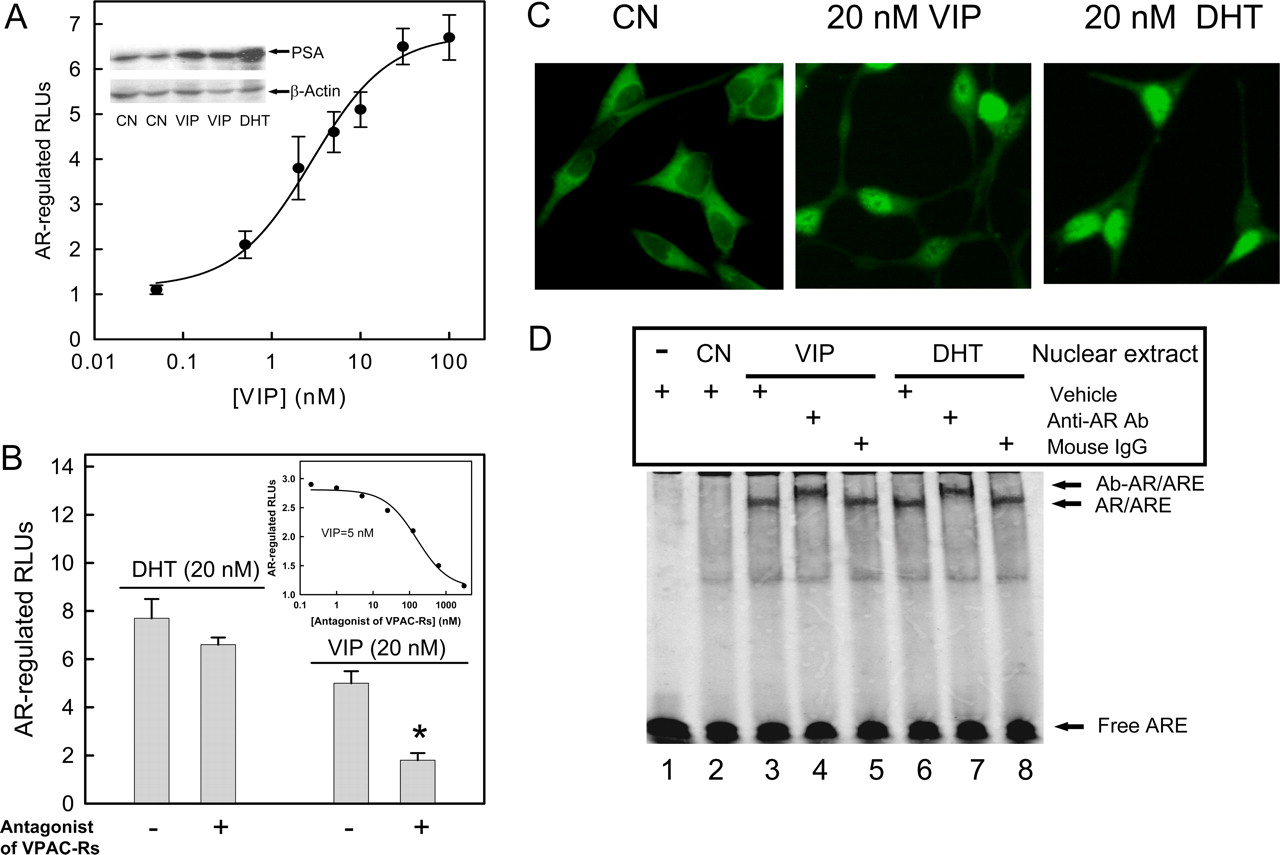
VIP-induced prostate cancer cell growth is dependent on VPAC-Rs and a functional AR. A, LNCaP cells were seeded in 96-well plates (5000 cells/well) and cultured in RPMI 1640 phenol red-free medium supplemented with 5% FBS (FBS) or 5% CSS without (CN) or with 20 nM VIP or DHT for 4 days. Total cell numbers in each group were counted at day 2 and 4. B, cells were cultured in steroid-depleted medium (5% CSS) without or with increasing concentrations of DHT or VIP for 48 h. C, cells were pretreated without (None) or with the VPAC-R antagonist [d-P-Cl-Phe(6)-Leu(17)]-VIP (2 μM) or the AR antagonist bicalutamide (20 μM) 30 min before treatment with 20 nM VIP or DHT for 48 h. In both B and C, the CellTiter 96 AQueous Cell Proliferation Assay (Promega) was used to determine the number of viable cells. The absorbance at 490 nm is directly proportional to the number of living cells. Absorbance before ligand treatment was subtracted from the data obtained. Data shown are means ± S.E. of triplicates, and these experiments were repeated at least five times. *, p < 0.01 compared with cells in the absence of antagonists (None). D, silencing AR by the AR-specific siRNA attenuates VIP or DHT-induced LNCaP cell growth. Cells cultured in steroid-depleted medium were seeded in 96-well plates and, 24 h later, were either mock-transfected or transfected with AR-specific siRNA or scrambled siRNA for 48 h. Cells were then treated without (CN) or with 20 nM DHT or VIP for 48 h, and total cell numbers in each well were counted. Cell growth is expressed as the percentage of the cells compared with the control wells (untreated mock-transfected cells). Data show the mean ± S.E. of three experiments, each with five replicates. *, p < 0.01 compared with control (CN). Inset, Western immunoblotting analysis of cell lysates from LNCaP cells mock-transfected (lane 1), cells transfected with scrambled siRNA (lane 2), and cells transfected with AR-specific siRNA (lane 3). Blot shows AR and β-actin protein expression.
Results
VIP Induces Androgen-Independent Growth of LN-CaP Cells. Because androgen-sensitive prostate cancer LN-CaP cells express VPAC-R, predominantly subtype 1 (Rekasi et al., 2000), we investigated the effect of VIP on the growth of LNCaP cells for its direct involvement in androgen-independent growth of prostate cancer cells. By directly counting changes of cell number, we found that the growth rate of LNCaP cells was dramatically reduced in androgen-depleted medium (5% CSS) (CN) compared with that in medium with 5% FBS. Addition of either androgen DHT (20 nM) or VIP (20 nM) into the androgen-depleted medium partially restored the LNCaP cell growth (Fig. 1A). Trypan blue exclusion analysis indicated that VIP or DHT did not change the viability of LNCaP cells. Under our experimental condition, LNCaP cell viability was approximately 90% in the presence and absence of ligands (data not shown). Thus, we further examined the effect of VIP on androgen-independent proliferation of LNCaP cells by means of the CellTiter 96 Aqueous One solution cell proliferation assay. As shown in Fig. 1B, VIP increased LNCaP cell proliferation in a dose-dependent manner (EC50 = 6.7 ± 2.4 nM) with a maximum of approximately 75% of that in the presence of an optimal concentration of DHT (20 nM). A 2 μM concentration of the VPAC-R antagonist [d-P-Cl-Phe(6)-Leu(17)]-VIP (Fizanne et al., 2004) blocked VIP-induced LNCaP cell proliferation, but not DHTstimulation (Fig. 1C), suggesting that VIP stimulates prostate cancer growth through VPAC-Rs in an androgen-independent manner, as seen in hormone-refractory prostate cancers.
VIP-Induced Prostate Cancer Cell Growth Is Dependent on a Functional AR. To demonstrate that AR signaling is required for VIP-induced androgen-independent proliferation of LNCaP cells, we used bicalutamide, a well-characterized AR antagonist (Maucher and von Angerer, 1993), to block AR function. As shown in Fig. 1C, preincubation of LNCaP cells with 20 μM bicalutamide completely blocked VIP- and DHT-induced cell proliferation. The antiproliferative effect of bicalutamide was not related to cytotoxicity or loss of viability. Trypan blue exclusion assay indicated 89.8 ± 1.7% viability after a 48-h exposure to 20 μM bicalutamide compared with 92.1 ± 0.5% in controls (n = 6); similarly, bicalutamide did not increase the proportion of detached cells (1.6 ± 0.5% compared with 1.1 ± 0.7% in controls). To further determine whether AR is still required for the VIP-stimulated prostate cancer cell growth, we performed AR protein expression knockdown using a synthesized AR-specific siRNA SMARTpool (Upstate). This siRNA pool suppressed over 75% of AR protein expression in LNCaP cells 48 h after transfection compared with cells transfected with a scrambled siRNA, as shown by immunoblot analysis (lane 3 versus 2 in Fig. 1D, inset). Cell growth assay indicated that DHT-dependent cell growth was attenuated by siRNA-mediated knockdown of the AR. In addition, VIP-stimulated androgen-independent cell growth was also abolished by the AR-specific siRNA but not the scrambled siRNA (Fig. 1D). Altogether, our results support the notion that VIP-induced androgen-independent cell growth in LNCaP cells requires a functional AR.
Androgen-Independent AR Activation by VIP. The effect of VIP on the transcriptional activity of AR was then examined using an AR-regulated reporter construct, ARE3-tk-LUC (Cao et al., 2006). The transfected cells were cultured in an androgen-depleted medium without or with exogenous VIP or DHT. Stimulation with VIP induced a dose-dependent increase in the AR-regulated reporter activity in LNCaP cells. The maximal response (7-fold) was achieved at a VIP concentration of 100 nM with an EC50 value of 3.0 ± 0.8 nM (n = 8). PSA is an AR-regulated serine protease secreted by the prostate. An elevation in the circulated PSA level is the most sensitive and reliable parameter for monitoring the relapse of prostate cancer after hormone therapies. Thus, we performed Western blot analysis to examine PSA expression in VIP-treated and untreated LNCaP cells using an anti-PSA antibody. Indeed, AR-regulated endogenous PSA expression in LNCaP cells was increased by 2.5 ± 0.7-fold (n = 6) after 20 nM VIP treatment (Fig. 2A, inset). As expected, 20 nM DHT increased AR-regulated luciferase activity and PSA expression by approximately 8- and 4.5-fold, respectively.
Congruent with its effects on VIP-stimulated LNCaP cell growth, 2 μM[d-P-Cl-Phe(6)-Leu(17)]-VIP blocked AR activity stimulated by 20 nM VIP but not by DHT (Fig. 2B). A typical competition curve with an IC50 value of 157 ± 42 nM was observed (Fig. 2B, inset) for this antagonist when it was titrated into an AR-regulated luciferase assay in the presence of 5 nM VIP.
Decreased expression of NEP, a cell surface neutral peptidase that degrades neuropeptide growth factors, is associated with prostate cancer progression (Papandreou et al., 1998). To test whether the NEP is involved in the VIP-induced AR transactivation, we examined 5 nM VIP-induced, AR-regulated luciferase activity in LNCaP cells cultured in steroid-depleted medium with or without 10 μM concentration of the NEP inhibitor phosphoramidon. Our result indicated that phosphoramidon had no significant effect on VIP-induced AR activation (data not shown), presumably because NEP activity in prostate cancer cells becomes almost negligible with androgen withdrawal (Papandreou et al., 1998).
VIP Increases the Nuclear Translocation of AR and Binding to ARE. Ligand-free AR is sequestered in the cytoplasm of prostate cancer cells. Upon androgen binding, AR dimerizes and translocates to the nucleus to bind specific DNA sequences, termed ARE, to activate the transcription of AR-regulated genes (Heinlein and Chang, 2004). We examined the subcellular localization of AR after agonist treatment. Both VIP and DHT caused the movement of AR into the nucleus of LNCaP cells (Fig. 2C). To determine whether VIP increased the DNA binding activity of the AR protein to the ARE, we performed EMSA using fluorescent-labeled oligonucleotides of the ARE. Nuclear extracts prepared from LNCaP cells treated with or without VIP or DHT were incubated with the IRDye-labeled double-stranded ARE. As shown in Fig. 2D, an increase in AR-ARE complex formation was evident with 20 nM VIP treatment compared with the untreated control (lane 3 versus 2). The specificity of the AR-ARE complex was demonstrated by its supershift with a monoclonal anti-AR antibody (lane 4 versus 3) but not control mouse nonimmune IgG (lane 5 versus 3). As a positive control, AR-ARE complex formation was significantly increased in the nuclear extract from LNCaP cells treated with 20 nM DHT (lane 6 versus 2), which was also supershifted by the anti-AR antibody (lane 7 versus 6) but not the control mouse IgG (lane 8 versus 6). Thus, VIP can activate AR for its translocation into the nucleus.
VIP Stimulates AR in Other AR-Positive Prostate Cancer Cell Lines. The CWR22Rv1 cell line, derived originally from a human primary prostate tumor, exhibits AR expression and PSA secretion. Its growth is androgen-responsive but with elements of androgen-insensitivity (Lee et al., 2001). In the absence of androgen, CWR22Rv1 cells were found to possess high basal AR activity as shown in reporter gene assays, implying an activated AR (Fig. 3A). Nevertheless, VIP treatment of CWR22Rv1 cells grown in steroiddepleted medium resulted in a further dose-dependent AR activation with a maximum stimulation of 1.8-fold. The addition of 2 μM VIP antagonist [d-P-Cl-Phe(6)-Leu(17)]-VIP suppressed the basal AR activity and also caused a parallel rightward shift in the dose-response curve. The EC50 value of VIP was increased from 25 to 385 nM (Fig. 3A), demonstrating that this stimulation was VPAC-R-specific. In contrast, the AR agonist DHT (20 nM) stimulated AR in CWR22Rv1 cells by approximately 2.6-fold, which was only slightly inhibited by [d-P-Cl-Phe(6)-Leu(17)]-VIP (data not shown).
Both LNCaP and CWR22Rv1 cells express mutated AR. To determine whether this VIP-dependent activation is a consequence of the AR mutation, we examined the regulation of wild-type AR by VIP in PC3 cells, an androgen-independent prostate cancer cell line that lacks expression of AR and functional VPAC-Rs (Gkonos et al., 2000). PC3 cells were transfected with AR-regulated reporter ARE3-tk-LUC plasmid, an AR expression plasmid and/or a VPAC2-R expression plasmid. As shown in Fig. 3B, AR-regulated luciferase activity was very low in PC3 cells transfected with only ARE3-tk-LUC, even in the presence of VIP or DHT. When PC3 cells were transfected with all three of these plasmids, AR-regulated luciferase activity was increased approximately 2-fold by 100 nM VIP; absence of either the VPAC2-R or AR plasmid eliminated this increase. In contrast, AR-regulated luciferase activity was induced approximately 3-fold by DHT (20 nM) in VPAC-Rs-deficient PC3 cells but not in AR-deficient PC3 cells. These findings demonstrate that VIP can activate wildtype AR and that both VPAC-R and AR are required for the VIP-induced, AR-regulated transcriptional process in prostate cancer cells. Because LNCaP cells express endogenous AR and VPAC-R and a greater stimulatory effect of VIP on AR was observed in this cell line, we continued to use this cell line as a model system to investigate VIP-stimulated signaling events that may interact with the AR pathway in prostate cancer.

Androgen-independent AR activation in LNCaP cells by VIP. Cells were cultured in steroid-depleted medium without (CN) or with DHT (20 nM) or various concentrations of VIP for 18 h. A, VIP stimulated AR-regulated luciferase reporter expression. Six hours before DHT or VIP treatment, dual reporter genes (ARE3-tk-LUC and pRL-tk) were transfected into LNCaP cells. Luciferase activities of cell lysates were measured using the dual luciferase assay system (Promega). Data show the mean ± S.E. of the normalized luciferase activities (n = 5) (RLU). Inset, VIP increased the expression of endogenous PSA protein. Cell lysates was analyzed by Western blotting using the anti-PSA antibody with β-actin as loading controls (inset is representative of six experiments). Each lane contained 30 μg of cell lysate. B, the VPAC-R antagonist [d-P-Cl-Phe(6)-Leu(17)]-VIP (2 μM) attenuated activation of AR by 20 nM VIP but not DHT. Data show the mean ± S.E. *, p < 0.01 (n = 4) compared with LNCaP cells in the absence of the VPAC-R antagonist (-). Inset, antagonist competition; 1:5 serial dilutions of the VPAC-R antagonist were incubated with LNCaP cells for 10 min before treatment with 5 nM VIP for 18 h. The data shown are the average of duplicates from a single experiment and are representative of two independent measurements. The solid lines are the fits of the data with an IC50 value of 157 ± 42 nM. C, subcellular localization of endogenous AR in LNCaP cells was determined by immunofluorescence. Representative immunofluorescent images are shown. D, EMSA results of AR binding to ARE induced by 20 nM VIP (lane 3 versus lane 2). The antibody (Ab) against AR (lane 4) but not the mouse IgG control antibody (lane 5) supershifted the AR/ARE complex. DHT (20 nM) also increased AR/ARE complex formation (lane 6 versus lane 2), which was supershifted by the AR antibody (lane 7 versus lane 6) but not mouse IgG (lane 8 versus lane 6).
Role of PKA and MAPK/ERK in VIP-Stimulated Activation of AR. It has been suggested that phosphorylation is involved in androgen-independent AR activation (Rochette-Egly, 2003). To assess whether phosphorylation plays a key role in VIP-stimulated AR activation, we examined the effect of various kinase inhibitors on AR-regulated reporter gene activation. Our initial study was focused on PKA, a major down-stream effector of Gs-coupled VPAC-Rs. Using a PepTag Nonradioactive PKA assay, we found that a 30 μM concentration of the PKA inhibitor H89 was needed for the complete inhibition of PKA activation after stimulation of LNCaP cells with 20 nM VIP (Fig. 4A, inset), consistent with previous observations that 30 μM H89 was needed to block PKA activation by isoproterenol in LNCaP cells (Kasbohm et al., 2005). A similar treatment of the LNCaP cells with H89 resulted in the dose-dependent reduction of VIP-induced AR transcriptional activation with the maximum inhibition of 80 ± 5% (Fig. 4A). We used PKI, a selective peptide inhibitor of PKA, to further investigate the role of PKA in VIP-induced AR activation. As shown in Fig. 4B, 10 μM PKI attenuated VIP-dependent AR activation by more than 70%, similar to the inhibitory effect of 30 μM H89. Taken together, these data indicate that VIP-induced AR activation in LNCaP cells is PKA-dependent.

Effects of VIP on AR-regulated luciferase transcription in prostate cancer CWR22Rv1 and PC3 cells. A, CWR22rv1 cells were transiently transfected with the dual reporter genes (ARE3-tk-LUC and pRL-tk) and then treated with various concentrations of VIP in the presence or absence of the VPAC-R antagonist [d-P-Cl-Phe(6)-Leu(17)]-VIP (2 μM) for 18 h in steroid-depleted medium. The basal AR activity without VIP-stimulation was 5.3 ± 0.2 and 4.2 ± 0.1 RLUs in the absence and presence of the VPAC-R antagonist, respectively. B, PC3 cells (24-well) were transfected with the dual reporter genes along with empty vectors or AR plasmid (100 ng) and/or VAPC2-R plasmid (400 ng) as indicated. After transfection, cells were treated without (None) or with either VIP (100 nM) or DHT (20 nM) for 18 h in steroid-depleted medium. The results are taken from three independent experiments with *, p < 0.01 compared with cells in the absence of agonists (None). Inset, PC3 cell lysates were analyzed for the expression levels of AR, VAPC2-R, and β-actin by Western blot assays. Lane 1, control; 2, transfected with AR; 3, transfected with VPAC2-R; 4, transfected with both AR and VPAC2-R.
We then tested inhibitors at doses that are selective for PI3K-Akt, PKC, Src, and MAPK/ERK signaling pathways, which all have been shown to be involved in androgen-independent AR activation (Rochette-Egly, 2003). As shown in Fig. 4B, neither the PKC inhibitor GF109203X (GF, 10 μM) nor the PI3K-Akt signaling inhibitor Ly294002 (Ly, 5 μM) showed any significant effect on the AR transcriptional activation by VIP. However, treatment with the MEK inhibitor U0126 caused a dose-dependent reduction in VIP-stimulated AR transcriptional activation with a maximum inhibition of 60 ± 4% (Fig. 4, A and B). In contrast, DHT-induced activation of AR was fully functional in the presence of 10 μM PKI or U0126 (Fig. 4B). It is interesting that the Src kinase inhibitor PP2 (10 μM) also inhibited VIP-induced AR activation by approximately 60%, suggesting a role for Src in signal flow from VPAC-Rs to AR in LNCaP cells.
To further demonstrate the important roles of PKA, Src, and MAPK/ERK in VIP-induced AR activation, we examined the effects of different inhibitors on VIP-induced, AR-regulated PSA up-regulation in LNCaP cells by Western blot analysis. As shown in Fig. 4B (inset), 20 nM VIP increased PSA protein level in LNCaP cells by approximately 3-fold (n = 3), which was attenuated by pretreatment of cells with 10 μM concentrations of PKI, U0126, and PP2 but not GF (10 μM) or Ly (5 μM).
Inhibition of PKA Abolished VIP-Induced ERK1/2 Activation. Previous studies have shown that MAPKs, particularly ERK1/2, are involved in androgen-independent AR activation in prostate cancer cells (Cao et al., 2006). It is interesting that VIP activated ERK1/2 in pituitary cells (Fernandez et al., 2005) and potentiated the ability of EGF, interleukin 6, and serum to activate MAPK in LNCaP cells (Chen et al., 1999). Thus, we examined ERK activity in LNCaP cells after VIP treatment through the measurement of ERK-activated, Elk-1-dependent expression of a luciferase reporter gene (Cao et al., 2006). As shown in Fig. 4C, treatment of LNCaP cells with VIP (20 nM) stimulated the ERK-regulated luciferase activity by approximately 6-fold, which was blocked by pretreatment with PKA inhibitor PKI (10 μM), Src inhibitor PP2 (10 μM), or MEK inhibitor U0126 (10 μM). The PKC inhibitor GF109203X (10 μM) and PI3K-Akt inhibitor Ly294002 (5 μM) had only minor inhibitory effects on VIP-induced ERK-regulated luciferase activity in LNCaP cells.
We further tested the ability of VIP to activate ERK1/2 in LNCaP cells by measuring the phosphorylation status of ERK1/2. In androgen-sensitive LNCaP cells, ERK1/2 is initially unphosphorylated but becomes phosphorylated in response to stimulation with VIP (Fig. 4C, inset). Phosphorylation of ERK1/2 in response to VIP was abolished by pretreatment of cells with 10 μM PKI or PP2. In contrast, 10 μM GF109203X or 5 μM Ly294002 had little effect on VIP-stimulated ERK1/2 activation. As a control, 10 μM U0126 completely abolished VIP-stimulated ERK1/2 phosphorylation.
As additional controls, we observed that the general PKC inhibitor GF (10 μM) effectively blocked PKC activation induced by 10 μM phenylephrine, an agonist of Gq-coupled α1A-adrenergic receptors, in a PepTag nonradioactive PKC assay (Promega), and 5 μM Ly294002 completely abolished 5% FBS-induced Akt phosphorylation in LNCaP cells as determined by Western blot analysis (data not shown). These results therefore exclude the possibility that the inability of 10 μM GF109203X or 5 μM Ly294002 to inhibit VIP-induced activation of ERK1/2 and AR was due to a failure of these inhibitors to block PKC and Akt activity, respectively, in LNCaP cells.
Blocking ERK1/2 Activation Attenuated VIP-Induced AR Activation. We used the dominant-negative mutants MEK1-K97M and ERK2-K52R (Cao et al., 2006) to inhibit endogenous ERK1 and ERK2 activity, respectively. Indeed, cotransfection of MEK1-K97M and ERK2-K52R inhibited VIP-induced ERK-regulated luciferase activity as shown in Fig. 5A, inset. In addition, the combination of MEK1-K97M plus ERK2-K52R (1:1 molar ratio) also caused a dose-dependent reduction in the VIP-stimulated AR-regulated luciferase activity with a maximum inhibition of 67 ± 5% (n = 4) (Fig. 5A), consistent with the maximal inhibition of VIP-dependent AR activation by U0126 (Fig. 4A). In contrast, transfection of these two plasmids had little effect on DHT-induced AR-regulated luciferase activity (Fig. 5A).

Blocking PKA-dependent ERK1/2 activation attenuated VIP-induced AR activation in LNCaP cells. LNCaP cells were transfected with the AR-regulated luciferase vector for 6 h. Cells were then treated with VIP (20 nM), DHT (20 nM), or vehicle in the presence or absence of inhibitors that were added 30 min before agonist treatment. Cells were analyzed for AR activity 18 h later by the dual luciferase assay (Promega). A, dose-dependent inhibition of VIP-induced AR activation by the PKA inhibitor H89 or the MEK inhibitor U0126. Inset, agarose gel exemplifying the effects of H89 on the VIP-induced PKA activation in LNCaP cells. PKA activity was examined in cell lysates using a PepTag assay for nonradioactive detection of PKA, and the samples were separated on a 0.8% agarose gel; 0 (negative) and 5 ng of activated PKAα (positive) was used as assay controls. B, effect of inhibitors on VIP- or DHT-induced AR activation in LNCaP cells. The PKA inhibitor PKI (10 μM), the PKC inhibitor GF (10 μM), the MEK inhibitor U0126 (10 μM), the Src inhibitor PP2 (10 μM) or the PI3K-Akt inhibitor Ly (5 μM) was used. Bars represent mean ± S.E. (n = 4; *, p < 0.01). Inset, LNCaP cells pretreated with different inhibitors were stimulated with or without 20 nM VIP for 24 h. Cell lysates were analyzed for the expression levels of PSA and β-actin by Western blot assays. C, LNCaP cells were transfected with ERK reporter plasmids for 18 h. Cells were then treated with 20 nM VIP, 20 nM DHT, or vehicle in the presence or absence of inhibitors as indicated for 6 h. RLUs were measured to assess ERK-regulated reporter gene expression as described previously (Cao et al., 2006). Inset, LNCaP cells were treated with 20 nM VIP or vehicle (CN) for 15 min in the absence (-) or presence of inhibitors as indicated. The levels of phosphorylated (pERK1/2) and total ERK1/2 were examined by Western blot analysis.
Constitutively Activated ERK2 Activates AR in the Absence of Androgen. To test whether active ERKs are sufficient for androgen-independent AR activation, we transfected LNCaP cells with vectors encoding a Myc-tagged wildtype ERK2-MEK1 fusion protein or a constitutively activated ERK2-MEK1-LA fusion protein (Xu et al., 2001). As shown in Fig. 5B, expression of ERK2-MEK1-LA mutant but not wild-type ERK2-MEK1 induced a dose-dependent increase in the AR-regulated gene transcription in the absence of androgen, with a maximum stimulation of approximately 3-fold compared with 6- and 8-fold stimulation of AR by 20 nM VIP and DHT, respectively.
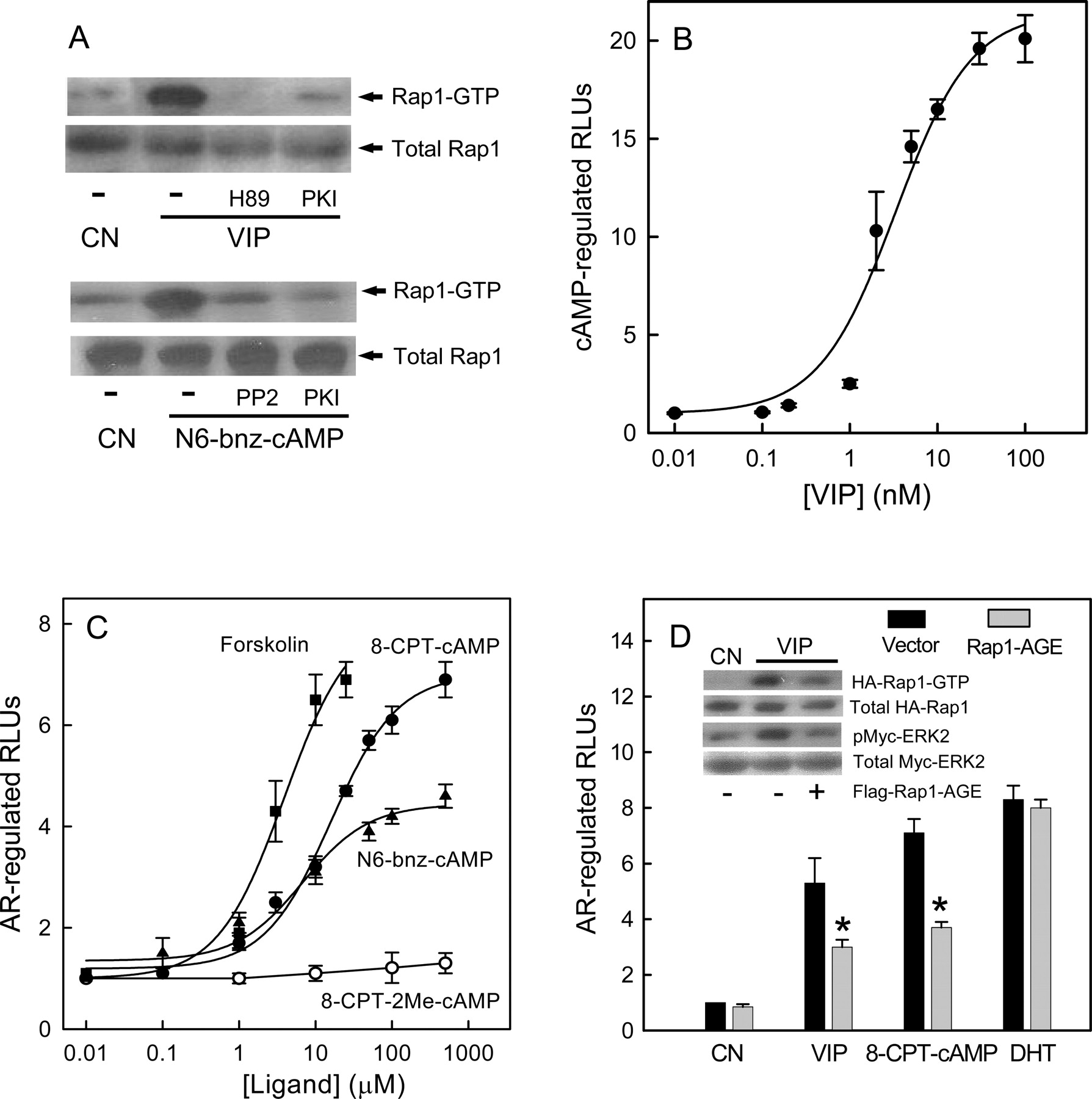
PKA-Dependent Activation of Rap1 Contributes to VIP-Induced AR Activation. It has been shown that small G-protein Rap1 plays a key role in hormonal activation of ERKs via Gs-coupled receptors in a number of systems, including those involving isoproterenol in S49 mouse lymphoma cells (Wan and Huang, 1998), thyroid-stimulating hormone in FRTL-5 rat thyroid cells (Iacovelli et al., 2001), and VIP in pituitary cells (Fernandez et al., 2005). However, the role of PKA in Rap1 activation remains controversial and may be cell-dependent, as shown in those studies. Thus, we examined the ability of VIP to activate Rap1 in LNCaP cells in the presence and absence of PKA inhibitors. As shown in Fig. 6A (top), 20 nM VIP significantly activated Rap1 in LNCaP cells, which was blocked by PKA inhibitors H89 or PKI, indicating that VIP-stimulated Rap1 activation in LN-CaP cells is PKA-dependent. Treatment with the PKA-specific activator N6-bnz-cAMP (50 μM) also resulted in Rap1 activation in LNCaP cells, which was blocked by 10 μM PKI (Fig. 6A, bottom). Last, we found that the Src tyrosine kinase inhibitor PP2 (10 μM) attenuated Rap1 activation induced by N6-bnz-cAMP, similar to the previous observation in NIH3T3 fibroblast cells that PKA activation of Rap1 is dependent on the Src tyrosine kinase (Schmitt and Stork, 2002).
Rap1 activation by Gs-coupled receptors is dependent on intracellular cAMP (Bos, 2003). To test the effect of VIP on cAMP production in LNCaP cells, LNCaP cells were transfected with the firefly luciferase reporter gene construct containing six copies of the CRE (Buhlmann et al., 1999). As shown in Fig. 6B, VIP generated a dose-response curve with a maximum stimulation of 20-fold in luciferase gene expression and EC50 values of 3.3 ± 1.0 nM, similar to the effect of VIP on androgen-independent AR activity (Fig. 2A). To demonstrate the importance of cAMP production in androgen-independent AR activation, we treated LNCaP cells with forskolin, a strong activator of adenylate cyclase. As expected, forskolin induced a dose-dependent increase in AR activation with a response of approximately 7-fold at 10 μM (Fig. 6C), further supporting the notion that cAMP production results in androgen-independent AR activation.

ERK1/2 are involved in VIP-mediated regulation of AR in LNCaP cells. LNCaP cells were transfected with dual reporter genes for AR activation along with a total of 2 μg of plasmids containing a control vector and various amounts of vectors encoding dominant-negative MEK1-KM and ERK2-KR mutants (molar ratio = 1:1) (A) or vectors encoding a wild-type Myc-tagged ERK2-MEK1 fusion protein or a constitutively activated Myc-tagged ERK2-MEK1-LA fusion protein (B). Cells were then treated without or with 20 nM VIP or DHT for 18 h and AR-regulated luciferase activities were measured. Data (mean ± S.E.) are shown for at least four independent experiments. *, p < 0.01 compared with cells transfected with control vectors. A, inset, transfection of 2 μg of dominant-negative MEK1-KM and ERK2-KR mutants (molar ratio = 1:1) blocked VIP-stimulated ERK-regulated luciferase activity. B, Western blot analysis of Myc-tagged fusion protein expression using an anti-Myc antibody, with lanes corresponding to the bars in the figure.
cAMP activates Rap1 via both PKA-dependent and -independent mechanisms. PKA-independent activation of Rap1 is believed to proceed via the direct activation of cAMP-responsive Rap1GEFs called EPACs (Bos, 2003; Wang et al., 2006). To directly establish the role of PKA-dependent and -independent activation of Rap1 in VIP-dependent AR activation, cell-permeable and nonhydrolyzable cAMP analogs selective for PKA and/or EPACs (Christensen et al., 2003) were used in our studies. We found that N6-bnz-cAMP, a cAMP analog that activates PKA but not EPACs, transactivated AR with a maximum response of approximately 4.5-fold and an EC50 value of 7.0 ± 3.0 μM, whereas 8-CPT-2Me-cAMP, a cAMP analog that retains a high affinity for EPACs but does not activate PKA, failed to activate AR in LNCaP cells even at a concentration of 500 μM (Fig. 6C). 8-CPT-cAMP, which activates both PKA and EPACs, transactivated AR with a maximum response of approximately 7-fold and an EC50 value of 15 ± 4.0 μM.
To further assess the involvement of PKA-dependent Rap1 activation in VIP-induced AR activation in LNCaP cells, we used Rap1-AGE, a Rap1 dominant-negative mutant that selectively blocks PKA-dependent Rap1 activation (Shi et al., 2004). Initial experiments confirmed that Rap1-AGE inhibited Rap1 activation by VIP. In addition, expression of Rap1-AGE in LNCaP cells also blocked VIP-induced ERK2 activation (Fig. 6D, inset), further supporting the role of Rap1 in VIP-induced ERK activation in LNCaP cells. As expected, expression of Rap1-AGE in LNCaP cells partially blocked AR activation by VIP or 8-CPT-cAMP (Fig. 6D) but not DHT.
Rap1GAP Attenuated VIP-Stimulated ERK and AR Activation. Rap1 is inactivated by a specific GTPase-activating protein (Rap1GAP). We investigated the effect of Rap1GAP on VIP-induced ERK activation by analyzing the level of active ERK1/2 (phosphorylated) in cell lysates. Rap1GAP plasmids were transfected into LNCaP cells with a Cell Line Nucleofector Kit V (Amaxa Biosystems) to achieve high transfection efficiency (approximately 70%). As shown in Fig. 7A, inset, VIP-treatment was associated with an induction of endogenous ERK1/2 phosphorylation that was partially blocked by transfected Rap1GAP. Incomplete inhibition of VIP-induced endogenous ERK1/2 activation by Rap1GAP is apparently due to partial transfection of LNCaP cells under our experimental conditions. LNCaP cells were then cotransfected with Rap1GAP and ERK-regulated luciferase reporter plasmids. ERK-regulated luciferase reporter gene assays demonstrated that recombinant Rap1GAP largely blocked VIP-stimulated ERK activation (Fig. 7A), suggesting that Rap1 activation is indeed required for VIP-dependent ERK activation. It should be noted that the inhibitory effect of Rap1GAP on VIP-stimulated ERK-regulated luciferase activity is specific because expression of Rap1GAP only slightly reduced EGF (5 ng/ml)-stimulated ERK-regulated luciferase activity in LNCaP cells (Fig. 7A), presumably because EGF activates ERKs in a Ras-dependent manner in LNCaP cells (Chen et al., 1999). DHT only had a modest stimulatory effect on ERK activity (approximately 1.7-fold) in LNCaP cells, which was not significantly affected by expression of Rap1GAP (Fig. 7A, inset).
Finally, we examined the change of VIP-induced AR activation after Rap1GAP expression in LNCaP cells. As expected, expression of Rap1GAP attenuated the VIP-dependent AR activation in a dose-dependent manner with a maximum inhibition of approximately 70% (Fig. 7B). It is interesting that the Rap1GAP or dominant-negative mutant Rap1-AGE failed to attenuate DHT-stimulated AR activation in LNCaP cells (Fig. 7, B and D), suggesting that a PKA-dependent activation of Rap1 only contributes to VIP-induced, but not DHT-stimulated, AR activation in prostate cancer LNCaP cells.
Discussion
The precise mechanisms underlying the conversion of prostate cancer to a state of androgen-independence (hormone refractoriness) remain poorly understood. In androgen-independent prostate cancer cells, AR is nevertheless activated even in the absence of androgens, and this activation remains crucial for androgen-independent prostate cancer proliferation (Zegarra-Moro et al., 2002). It is foreseeable that there would be a transition stage in which both androgen-dependent and -independent activation of AR occurs. Identification of factors and mechanisms by which these factors transactivate AR in androgen-sensitive prostate cancer cells should significantly advance our knowledge of prostate cancer progression. VIP, one of the relatively abundant neuropeptides in the human prostate, is a particularly interesting mediator of AR transactivation. Although VIP seems to act as both a growth factor and survival factor in androgen-independent prostate cancer cells, little is known about its role in transactivating AR and promoting androgen-independent growth of androgen-sensitive prostate cancer cells. In the present study, we conducted experiments to delineate a molecular mechanism used by androgen-sensitive LNCaP cells, the most studied model of human prostate cancer, when responding to VIP.

PKA-dependent activation of Rap1 contributes to VIP-induced AR activation. A, LNCaP cells were pretreated with inhibitors or vehicle(-) for 30 min and then incubated without (CN) or with 20 nM VIP or N6-bnz-cAMP (50 μM) for 10 min. Top, cells were pretreated with H89 (30 μM), PKI (10 μM), or vehicle(-). Bottom, cells were pretreated with PP2 (10 μM), PKI (10 μM), or vehicle(-). Cell lysates were then examined for activated Rap1 (Rap1-GTP) as described under Materials and Methods. Total Rap1 protein levels are also shown. B, concentration-dependent cAMP-regulated luciferase activity responses of VIP in LNCaP cells. Results are given as fold induction over basal activity. C, concentration-dependent AR activation by the adenylyl cyclase activator forskolin, the PKA-activator N6-bnz-cAMP, and 8-CPT-cAMP but not EPAC-selective activator 8-CPT-2Me-cAMP. D, Rap1 dominant-negative mutant Rap1-AGE attenuated AR activation by VIP or 8-CPT-cAMP but not DHT. LNCaP cells were transfected with dual AR luciferase reporter gene along with 1 μg of empty vector or vector encoding Rap1-AGE. Twenty-four hours after transfection, cells were treated without (CN) or with 20 nM VIP, 100 μM 8-CPT-cAMP, or 20 nM DHT for 24 h, and luciferase activities of cell lysates were then measured. Data (mean ± S.E.) are shown for two independent experiments each conducted in triplicate. *, p < 0.01 compared with cells transfected with control vectors. Inset, LNCaP cells were transfected with 9 μg of vector (-) or Flag-Rap1-AGE plasmid (+) along with 1 μg of HA-Rap1 or Myc-ERK2. Cells were then treated without (CN) or with 20 nM VIP for 10 min. Cell lysates were examined for activated HA-Rap1 (HA-Rap1-GTP). Total HA-Rap1 in cell lysates is indicated as a loading control. Myc-ERK2 was immunoprecipitated from cell lysates using an anti-Myc antibody and analyzed by immunoblotting with the phospho-ERK antibody (pMyc-ERK2). The levels of total Myc-ERK2 are indicated as a loading control.
We found that in the absence of androgens, VIP induced a dose-dependent increase in LNCaP cell growth, with a plateau value of approximately 75% of that seen for DHT responses at the optimal concentration. In the presence of VIP, the growth of androgen-sensitive LNCaP cells becomes androgen-independent but still requires a functional AR because the VIP response can be blocked by the AR antagonist bicalutamide or AR siRNA, consistent with the notion that AR still plays a crucial role in androgen-independent prostate cancer growth. We therefore verified that VIP activated AR. Immunocytochemistry analysis demonstrated that both DHT and VIP caused a redistribution of AR from the cytoplasm to the cell nucleus, an initial step for activated AR to regulate gene transcription. These results are further supported by the EMSA analysis showing that activated AR capable of forming complexes with ARE consensus oligonucleotides were present in nuclear extracts from LNCaP cells treated with VIP or DHT but not in nuclear extracts from unstimulated LNCaP cells. We also used both an AR-dependent luciferase reporter gene assay and Western blot analysis of AR-mediated stimulation of PSA expression to demonstrate that VIP indeed stimulated AR in the absence of androgen in LNCaP cells. Consistent with its inhibitory effect on androgen-independent cell growth induced by VIP, a VIP antagonist also blocked VIP but not DHT responses on AR activity, demonstrating different but converging sites of action for these two agonists on AR.

Rap1GAP attenuated both VIP-stimulated ERK and AR activation. LNCaP cells cultured in steroid-depleted medium were transfected with ERK-regulated (A) or AR-regulated (B) luciferase reporter plasmids along with 1 μg of empty vector or vector encoding GFP-tagged Rap1GAP. Cells treated without (CN) or with 20 nM VIP or 20 nM DHT for 6 h (A) or 18 h (B) then were harvested for luciferase assays. A, data (mean ± S.E.) are shown for three independent experiments. *, p < 0.01 compared with cells transfected with control vectors. Treatment of LNCaP cells with 5 ng/ml EGF for 6 h was used as a control and induced approximately a 16-fold stimulation of ERK-regulated luciferase activity. Inset, LNCaP cells were transfected with GFP-tagged Rap1GAP (+) or vector (-). Cells were then treated without (CN) or with 20 nM VIP or DHT for 15 min. Cell lysates were examined by immunoblotting with phospho-ERK1/2 antibody (pERK1/2) and total ERK1/2 antibody. B, data are shown as a percentage compared with cells transfected with control vectors, where 100% equals 6- and 9-fold stimulation of AR activity by 20 nM VIP and DHT, respectively. Expression of GFP had no effect on VIP-stimulated ERK and AR activation in LNCaP cells (data not shown). Inset, Western blot analysis using anti-GFP antibody with β-actin as a loading control.
VIP also induced activation of AR via VPAC-Rs in the CWR22Rv1 cell line, another AR-positive and androgen-responsive human prostate cancer cell line. More importantly, by using the androgen-independent prostate cancer PC3 cell line, which lacks expression of endogenous AR and VPAC-Rs, we demonstrated that VIP can activate transiently transfected wild-type AR and that both VPAC-Rs and AR are required for VIP-induced changes in the AR-dependent transcriptional process in PC3 cells. All of these results affirm that VIP-mediated transactivation of AR is not restricted to LNCaP cells.
VPAC-Rs are typically coupled to Gs proteins, with stimulation leading to cAMP generation and subsequent PKA activation in various tissues, including prostate cancer cells (Gutiérrez-Cañas et al., 2005). In fact, our results indicated that VIP stimulation of AR could be largely abolished by pretreatment of LNCaP cells with PKA inhibitors, suggesting a crucial role of PKA. The mechanisms by which PKA modulates AR are still unclear. Some studies suggested that PKA directly phosphorylates AR to regulate its activity (Zhou et al., 1995). PKA may indirectly regulate the nucleocytoplasmic shuttling of AR and thus control its responsiveness (Kasbohm et al., 2005). As an alternative, PKA activation can modulate the activation of the MAPK/ERK system in prostate cancer cells (Gutiérrez-Cañas et al., 2005). Active MAPK/ERKs can directly phosphorylate and activate AR in vitro (Yeh et al., 1999) and promote hormone-independent but AR-dependent growth of androgen-responsive prostate cancer cells (Gao et al., 2006). The data presented here demonstrated that ERK1/2 is activated by VIP treatment, which may be very important for prostate cancer progression because activated ERKs are often found in hormone-refractory prostate cancer (Gioeli et al., 1999) and contribute to androgen-independent AR activation in androgen-independent prostate cancer cells (Cao et al., 2006). Consistent with this hypothesis, we found that VIP-stimulated AR activation was dependent on ERK1/2 activation, and expression of constitutively activated ERK2 significantly increased androgen-independent activation of AR. In addition, PKA functions upstream of ERK1/2 in LNCaP cells, because VIP-dependent ERK1/2 activation was blocked by the PKA inhibitors.
One intermediary in the signal flow from VIP-induced cAMP production to AR via ERK1/2 is Rap1, a ubiquitously expressed Ras-like protein. Rap1 is activated by guanine exchange factors, including EPACs and C3G, and is inactivated by Rap1GAP. Our results demonstrated that expression of a recombinant Rap1GAP in LNCaP cells significantly attenuated VIP-induced ERK and AR activation, suggesting an important role of Rap1 activation in the VIP-triggered signal transduction. Whether Rap1 can activate ERKs is still somewhat controversial (Enserink et al., 2002; Stork and Schmitt, 2002) and may be dependent on the mode of Rap1 activation in cells. Activation of Rap1 by EPACs is PKA-independent and is limited to activation of a perinuclear pool of Rap1 that does not cause ERK activation (Bos, 2003; Wang et al., 2006). On the other hand, C3G induces PKA-dependent activation of Rap1 that is responsible for cAMP-dependent ERK activation (Gotoh et al., 1995; Wang et al., 2006). We found that treating LNCaP cells with VIP induced an increased level of active Rap1, which was abolished by PKA inhibitors H89 and PKI, demonstrating that the response is PKA-dependent. This was further supported by our result showing that the PKA-specific activator N6-bnz-cAMP also caused Rap1 activation in LNCaP cells, which was blocked by PKI. It is interesting that the dominant-negative Rap1-AGE mutant partially blocked AR stimulation by both VIP and 8-CPT-cAMP, consistent with previous data that the dominant-negative Rap1-AGE mutant inhibits PKA-dependent Rap1 activation by C3G (Shi et al., 2004). In addition, it has been suggested that activation of Src by PKA triggers the assembly of a complex of Src and C3G to activate Rap1 and ERKs (Schmitt and Stork, 2002), consistent with our data that Src inhibitor PP2 not only attenuated Rap1 activation but also blocked VIP-induced activation of ERK1/2 and AR in LNCaP cells.
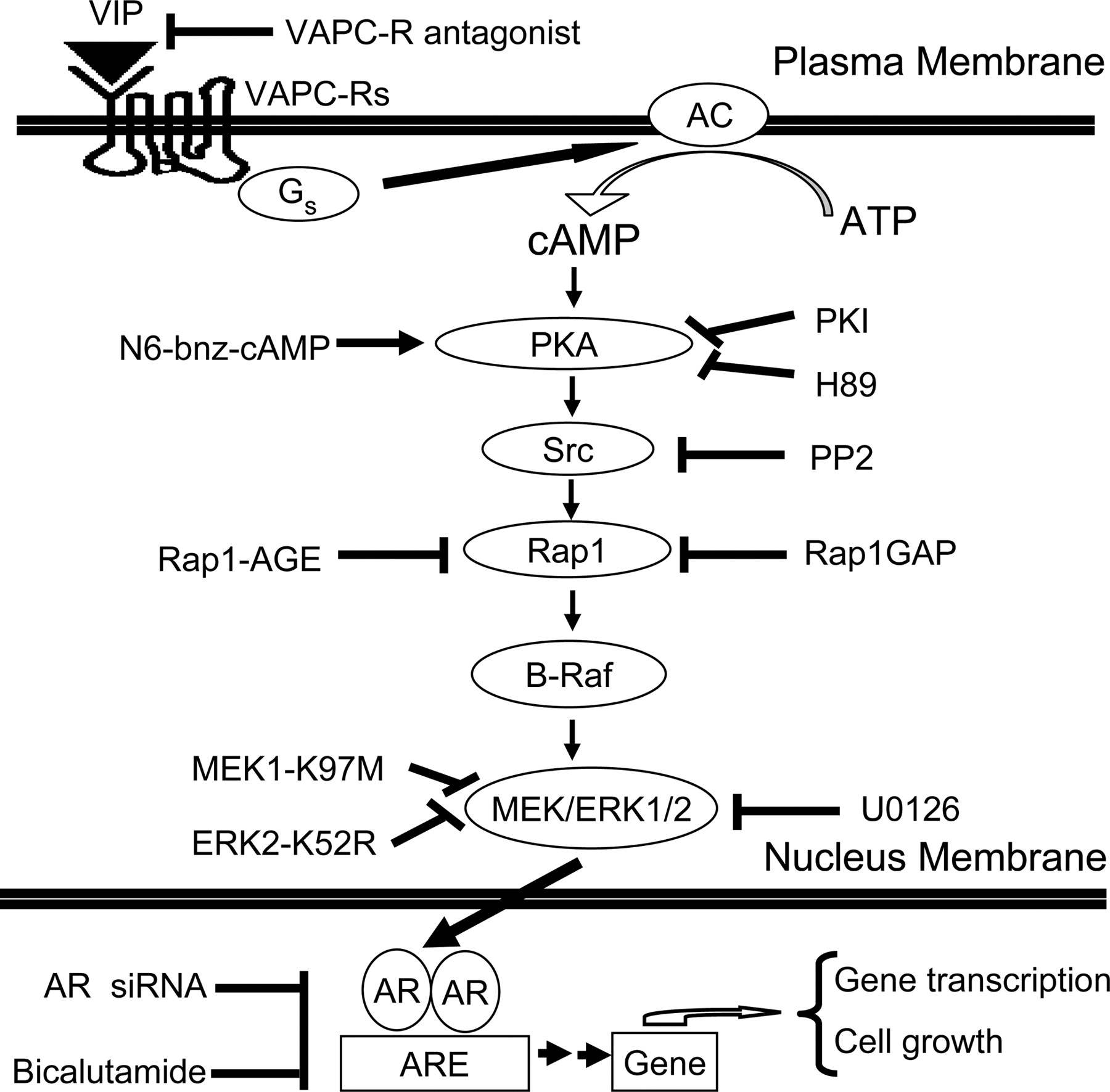
A model for signal relay from Gs-coupled VIP receptor to AR in androgen-sensitive prostate cancer cells. AC, adenylyl cyclase. The neuropeptide VIP induces androgen-independent AR activation via a signal relay from VPAC-Rs (blocked by the VPAC-R antagonist), Gs, cAMP, PKA (inhibited by H89 or PKI), Src (inhibited by PP2), Rap1 (blocked by Rap1GAP or dominant-negative mutant Rap1-AGE), B-Raf, and MEK/ERK1/2 (inhibited by U0126, dominant-negative mutants MEK1-K97M and ERK2-K52R). The PKA-specific activator N6-bnz-cAMP, but not the EPAC-selective activator 8-CPT-2Me-cAMP, transactivates AR. Transactivation of AR by VIP promotes androgen-independent growth of androgen-sensitive prostate cancer cells, which can be attenuated by AR-specific siRNA or the AR antagonist bicalutamide.
Daaka and his colleagues recently reported that Gs-coupled β2-adrenergic receptor signaling can activate AR responses in prostate cancer cells (Kasbohm et al., 2005). These authors also observed PKA-dependent AR activation with the β2-adrenergic receptor agonist isoproterenol. However, in that experiment, ERK1/2 was not activated in association with AR activation by isoproterenol. The reason for these differences is not currently clear. Either the LNCaP cells studied by Kasbohm et al. (2005) differ somehow from those studied by us, or some other experimental parameter is responsible.
In summary, our data are consistent with the model depicted in Fig. 8, where neuropeptide VIP induces androgen-independent AR activation via a signal relay from VPAC-Rs to AR in androgen-sensitive prostate cancer cells in a PKA/Rap1/ERK1/2-dependent manner. Transactivation of AR by VIP promotes androgen-independent growth of prostate cancer cells, which was attenuated by AR-specific siRNA or the AR antagonist bicalutamide. It should be noted that the transactivation of AR via VPAC-Rs is obviously very complicated, with a convergence of multiple signaling pathways. Our results have not eliminated the possibility of additional intermediate steps. For example, AR transactivation is probably a consequence of AR phosphorylation that occurs in a PKA- and ERK-dependent manner, but this requires further investigation. Experiments to further define the mechanisms leading to androgen-independent activation of AR are in progress.
Finally, our study may have clinical implications. For example, perineural invasion, characterized by a concentric ring of cancer cells around nerve fiber bundles, is associated with advanced prostate cancer, and exiting of cancer cells from the prostate gland via the perineural space may represent one of the major pathways for prostate cancer metastasis (Ayala et al., 2004). It is conceivable that prostate cancer cell invasion of the perineural space causes compression damage to the prostate nerves, leading to up-regulation of VIP generation in prostate nerves (Kashiba et al., 1992), which could then exert mitogenic effects on prostate cancer cells via VPAC-Rs despite androgen-deprivation therapies. It is interesting that the NEP that degrades neuropeptides is transcriptionally activated by androgen, and its activity is significantly reduced in prostate cancer cells with androgen withdrawal. Clinical evidence demonstrates that androgen deprivation plays a major role in the progression of prostate cancer to androgen independence. It is possible that downregulation of NEP under androgen-deprivation therapies facilitates the ability of extracellular neuropeptide growth factors such as VIP to bring about AR transactivation, thereby promoting the progression of prostate cancer. Thus, fully understanding the mechanisms that regulate VIP-stimulated transactivation of AR will lead to the better understanding of the pathogenesis of prostate cancer progression. More importantly, such studies are likely to reveal additional targets for treatment of hormone-refractory prostate cancers. For example, a VIP antagonist, which could potentially antagonize prostate cancer progression, may be a useful addition to the prostate cancer treatment formulary.
Acknowledgments
We thank Lyudmilla Batalkina for expert experimental assistance, Dr. Melanie H. Cobb (University of Texas Southwestern Medical Center) for providing various MEK1 and ERK2 plasmids, Dr. Hitoshi Kitayama (Kyoto University Graduate School of Medicine) for Flag-Rap1-AGE dominant-negative mutant, and Dr. A. Himmler (Boehringer Ingelheim Research and Development) for the pADneo2 C6-BGL plasmid. We also thank Dr. Keith R. Johnson and Dr. Frank Dowd for their helpful suggestions and review of the manuscript.
Footnotes
-
This work was supported in part by National Institutes of Health grant number P20-RR018759 from the National Center for Research Resources (to Y.T., M.F.L.), CA88184 (to M.F.L.), Nebraska State LB692 (to Y.T.), and American Cancer Society RSG-07-090-01-TBE (to Y.T.).
-
ABBREVIATIONS: AR, androgen receptor; ARE, androgen response element; 8-CPT-2Me-cAMP, 8-(4-chlorophenylthio)-2′-O-Me-cAMP; 8-CPT-cAMP, 8-(4-chlorophenylthio)-cAMP; N6-bnz-cAMP, N6-benzoyladenosine-cAMP; C3G, Crk SH3 domain guanine nucleotide exchanger; CRE, cAMP response element; CSS, charcoal-stripped fetal bovine serum; DHT, dihydrotestosterone; EGF, epidermal growth factor; EMSA, electrophoretic mobility shift assay; EPAC, exchange protein directly activated by cAMP; ERK, extracellular signal-regulated kinase; FBS, fetal bovine serum; GFP, green fluorescent protein; GPCR, G protein-coupled receptor; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase kinase; NEP, neutral endopeptidase; PBS, phosphate-buffered saline; PKA, cAMP-dependent protein kinase; PKI, myristoylated protein kinase inhibitor amide 14-22; PI3K, phosphoinositide-3 kinase; Rap1GAP, Rap1 GTPase-activating protein; Rap1GEF, Rap1 guanine nucleotide exchange factor; siRNA, small interfering RNA; VIP, vasoactive intestinal peptide; VPAC-R, vasoactive intestinal peptide receptor; PSA, prostate-specific antigen; HA, hemagglutinin; PKC, protein kinase C; RLU, relative luciferase activity units; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine; H89, N-[2-(4-bromocinnamylamino)ethyl]-5-isoquinoline; U0126, 1,4-diamino-2,3-dicyano-1,4-bis(methylthio)-butadiene; Ly294002, Ly, 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride; GF109203X, GF, 3-[1-[3-(dimethylaminopropyl]-1H-indol-3-yl]-4-(1H-indol-3-yl)-1H-pyrrole-2,5-dione monohydrochloride.
- Received January 4, 2007.
- Accepted April 12, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}