Article Text

Statistics from Altmetric.com
Is apoptosis the heart of the problem?
One of the “banes” of most health professionals who look after athletes and workers is tendinopathies. What causes them? What gets them better? Recently there have been several advances that may contribute to our understanding of these disorders.
We all know that tendinopathies occur in the tendinous portion of musculotendinous units that cross joints, often two joints—for example, the extensor carpi radialis brevis in tennis elbow, the patellar tendon in jumpers knee—and that they occur in situations of repetitive, high, often eccentric loading. The classic pathology is a loss of the normal collagenous architecture and replacement with an amorphous mucinous material that lacks the parallel, longitudinal architecture of normal tendon.1 Soslowsky et al2 made a major advance when they developed an animal model that could reproduce many of the microscopic changes of supraspinatus tendinopathy. The model involved running rats on a treadmill for up to an hour a day for 4–16 weeks. Supraspinatus tendons in the exercised animals had an increase in cellularity and loss of their normal collagen fibre organisation. The tendons were larger than normal in cross sectional area and had deteriorated mechanical properties. Specifically, they had a decreased modulus of elasticity and a decreased maximum stress at failure.
If over-exercise is so important, then the next question is how stress translates into these degenerative changes? Arnoczky et al3,4 have recently shown in cultured canine patellar tendon cells that there is a direct relation between the amount of stress the tendon cells see and the induction of a stress activated protein kinase (c-Jun N-terminal kinase (JNK)). Cyclic strain resulted in an immediate activation of JNK, which peaked at 30 minutes. This activation was regulated by a magnitude dependent, but not frequency dependent, calcium dependent mechanotransduction pathway. Whereas transient JNK activation is associated with normal cell processes, persistent JNK activation has been linked to the initiation of apoptosis—or programmed cell death.5
This work links nicely with some of our recent work,6,7 which shows that there is indeed increased apoptosis or programmed cell death in human rotator cuff tendons with tendinopathy. We compared apoptosis by using the TUNEL and DNA laddering techniques in the edges of torn rotator cuff supraspinatus tendons removed at surgery with normal appearing samples of subscapularis tendons also taken at surgery. There was a threefold increase in apoptosis in torn tendinopathic rotator cuff tendon compared with the control tendon. The proportion of cells undergoing apoptosis increased with age and was also greater in the “normal” subscapularis tendons of patients with torn rotator cuff tendons compared with subscapularis tendons of patients with intact supraspinatus tendons.
“ …there is indeed increased apoptosis or programmed cell death in human rotator cuff tendons with tendinopathy”
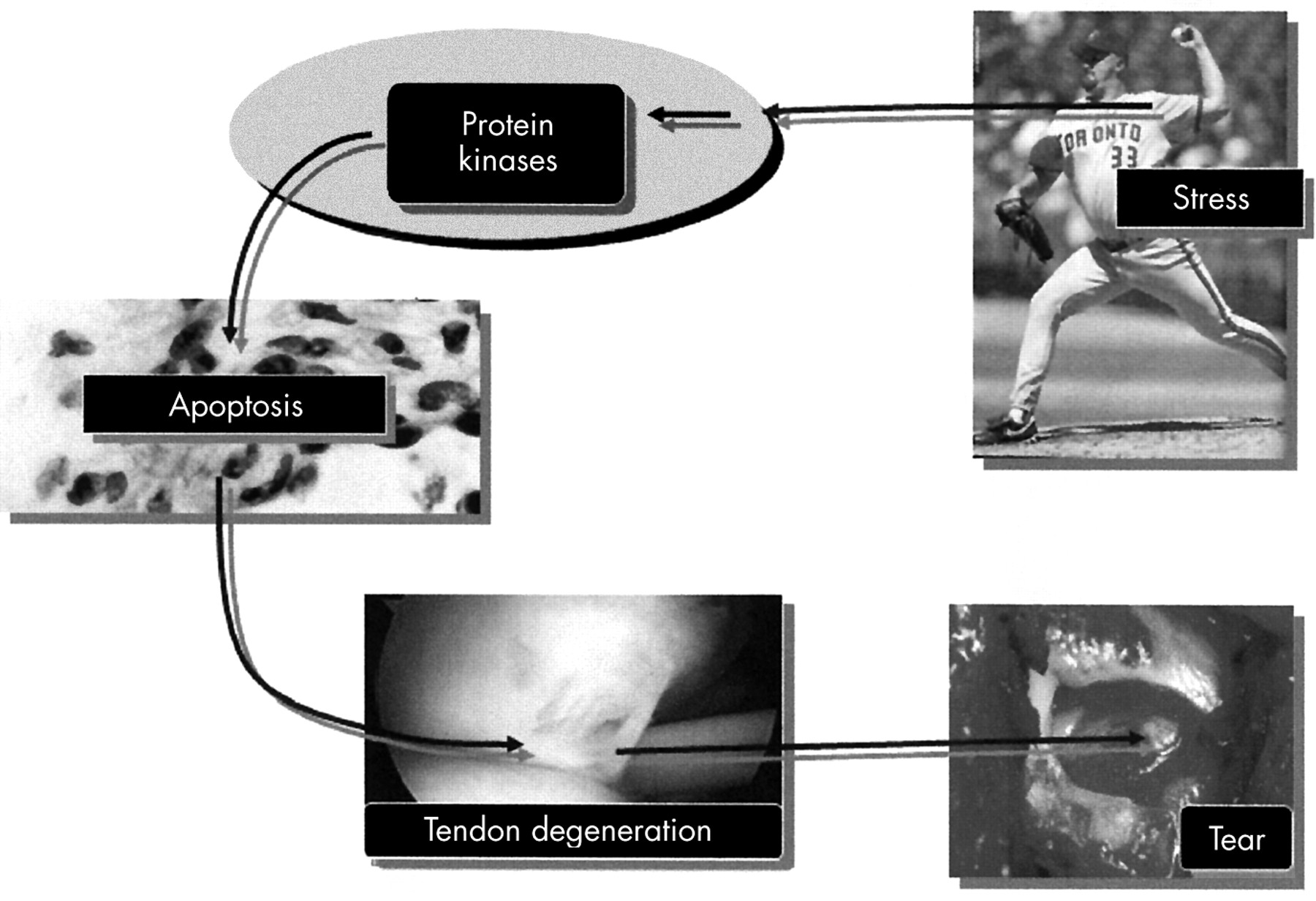
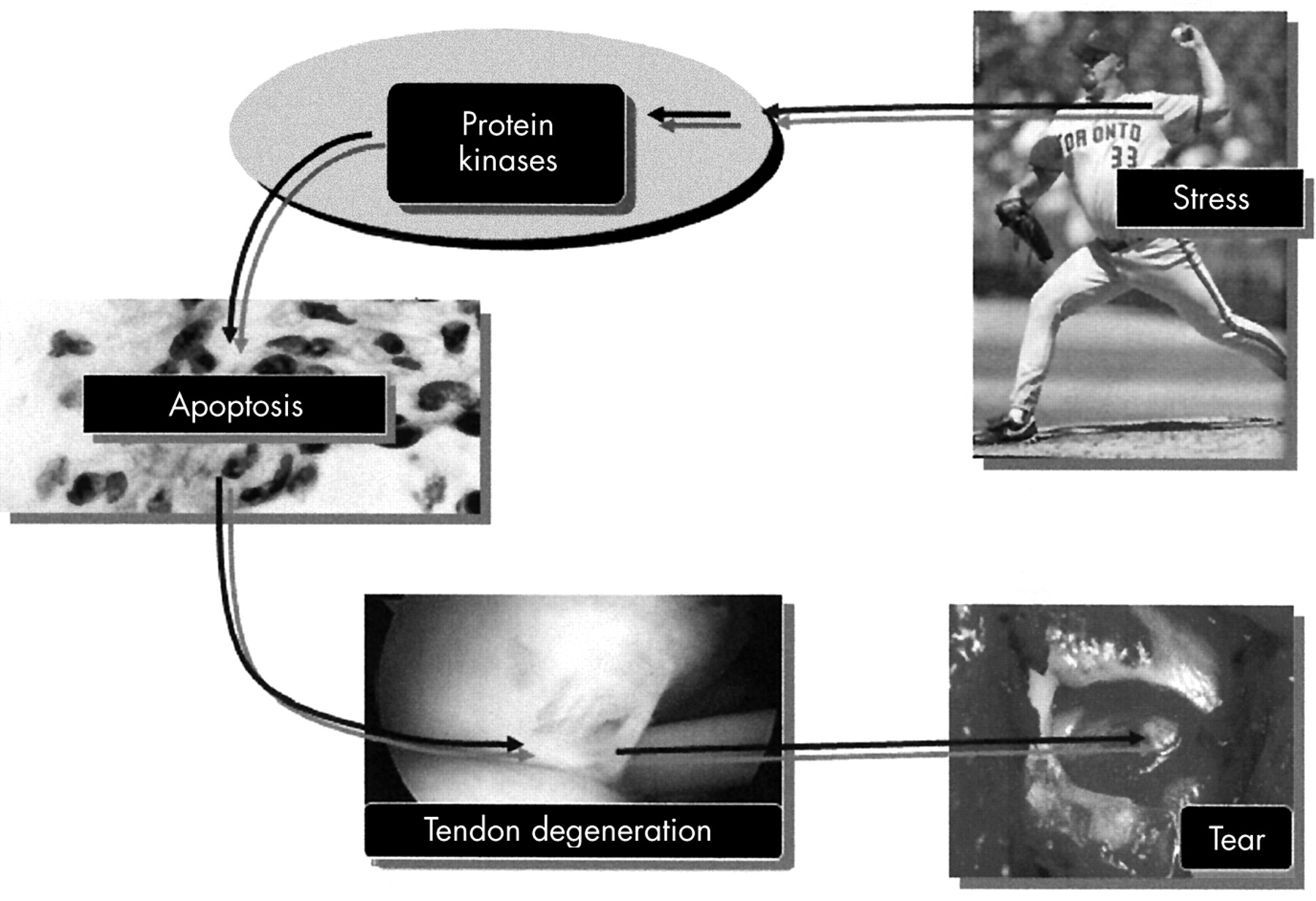
We can hypothesise therefore that tendinopathy may follow a pathway illustrated in fig 1. An increase in the amount and duration of load that a tendon cell sees may result in activation of intracellular stress activated protein kinases, which when persistently activated cause the tendon cells to undergo apoptosis or programmed cell death. Increased cell death results in a collagenous matrix which is weaker and more prone to tearing. With time, this tendon may rupture.
Clearly, there are many details to insert into this pathway but there is hope that if we can flesh out the fine details of the pathway, then we may be able to develop strategies to break the cycle at one or more points and prevent and/or treat tendinopathy more effectively.
{kind=link}
A schematic representation of how tendinopathies may arise. An increase in the amount and duration of load that a tendon cell experiences may result in activation of protein kinases, which when persistently activated cause the tendon cells to undergo apoptosis (programmed cell death). Increased cell death results in poor collagen synthesis and matrix remodelling and a collagenous matrix that is weaker and more prone to tearing. With time, this tendon may rupture.
Acknowledgments
This work was supported in part by South Eastern Sydney Area Health Service/St George Hospital and NiCox Corporation.
Is apoptosis the heart of the problem?