Article Text

Abstract
Athletic performance tests the limits of the human body and mind. Awe-inspiring achievements is what makes sports so fascinating. It is well appreciated however that top-level sports may sometimes overtax the body, and can lead to injuries, most notably of musculo-skeletal nature. This paper defends the thesis that the heart can also develop sports injuries at the ventricular level. We will elaborate on our hypothesis, originally put forward in 2003, that intense endurance activities put a particularly high strain on the right ventricle (RV), which over time, may lead to a proarrhythmic state resembling right (or less often) left ventricular cardiomyopathy. This can develop even in the absence of underlying demonstrable genetic abnormalities, probably just as a result of excessive RV wall stress during exercise. The syndrome of ‘exercise-induced arrhythmogenic RV cardiomyopathy’ may easily be overlooked. Sports cardiologists, like orthopaedic specialists, should be prepared to realise that excessive sports activity can lead to cardiac sports injuries in some, which will help to council on safe participation in all.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Ventricular arrhythmias in athletes are rare, but by nature they may be life threatening. Physical activity is associated by a 2.5 times increased risk for sudden death.1 The classical concept is that arrhythmic events are due to underlying (structural or electrical) heart disease, on which physical activity acts as a trigger for initiation of arrhythmias. Indeed, many autonomic and metabolic modulators are influenced by exercise, and may create the milieu for initiation and maintenance of ventricular arrhythmias. A multitude of underlying cardiovascular conditions have been shown to predispose an athlete to exercise-related sudden death in multiple registries.1,2
We presented a second, additional, hypothesis in 2003, postulating that intense endurance activities may lead to cardiac alterations due to a particularly high strain on the right ventricle (RV).3 Excessive RV strain may lead to minor cell damage after an endurance event, as evidenced by cardiac enzyme elevations that are very commonly observed after endurance competition. Over time, repetitive injury may lead to changes that resemble right (or less often) left ventricular (LV) cardiomyopathy, even in the absence of underlying demonstrable genetic abnormalities. The syndrome of ‘exercise-induced arrhythmogenic RV cardiomyopathy’ may have been overlooked and under-recognised in prior series. The aim of this article is to describe the pathophysiological findings that support the hypothesis of ‘exercise-induced arrhythmogenic right ventricular cardiomyopathy (ARVC)’.
Ventricular Arrhythmias In Athletes: Most Often A Right Ventricular Origin
Sudden death in athletes usually is the result of ventricular tachy-arrhythmias. The most common underlying cardiovascular abnormalities that have been identified in younger athletes (<35 year) are hypertrophic cardiomyopathy, coronary anomalies, ARVC, myocarditis and channelopathies, while silent coronary artery disease is the main cause in athletes over 35 years.1 ,2 Given this distribution of underlying disease, we were puzzled by findings in 46 high-level endurance athletes (performing ≥3 × 2 h of sports per week for more than 5 years; 80% competitive; 80% cyclists) that were referred to us for evaluation in the context of aspecific symptoms like palpitations and dizziness, but that could later be attributed through work-up to ventricular arrhythmias.3 The great majority of those ventricular arrhythmias (86%) had a RV origin (ie, with left bundle branch block morphology in the right precordial leads), which would not be expected based on the mentioned underlying pathologies, which statistically would have a much higher probability of inducing arrhythmias of LV origin. Nevertheless, the arrhythmic outcome, although clinically mild at presentation, proved to have an ominous course: after a medium follow-up of 4.7 years, 18 out of 46 had a major rhythm disorder, of which 9 were (aborted) sudden deaths (all cyclists; a mean of 3 years after presentation). Only an electrophysiological study inducing re-entrant arrhythmias was predictive for later arrhythmic events outcome (RR 3.4; p = 0.02), pointing to an underlying structural substrate. Although the athletes presented with RV arrhythmias, overt structural findings of ARVC were less frequently present than seen in familial forms. Only 1/46 had a familial history. Nevertheless, many of them showed other electrical signs of right ventricular changes like deep negative T-waves in the right precordial leads up to or even beyond V3, or the presence of late potentials on the signal averaged ECG (each about 40%).3 Combining major and minor criteria of the (original) ARVC diagnostic framework, 59% had manifest ARVC and an additional 30% had probable ARVC.4 It is of interest that others have also noted that asymptomatic athletes, and especially endurance athletes, develop changes on the signal averaged ECG, intermediate to those of controls and those of athletes with ventricular arrhythmias.5 We therefore wondered in how far structural adaptation of the athlete's RV, especially under extreme endurance load, had developed into RV degeneration and arrhythmogenicity.3
Many studies have shown that in athletes performing a marathon, triathlon or other ultra-endurance event, there are increases in B-type natriuretic peptide (BNP; in 54 to 100%), and cardiac enzymes like CK-MB, troponin-T and troponin-I (in 40–57%).6–13 The rise in cardiac enzymes is somewhat less in subjects performing habitual exercise,6 but cannot be denied even when experienced athletes perform high-intensity endurance events. However, echocardiographic studies have shown that the LV at the end of such an event usually has dimensions comparable with those before the event, with preserved, or minimally altered, global and regional function,6 ,7 ,13–20 Given the minimal alterations in LV measures, no correlation was noted between the enzyme rise and the degree of LV dilatation/hypokinesia.21 ,22 In contrast, RV dilatation and global hypokinesia are noted in at least one third of the athletes.6 ,7 ,13–15 ,17 ,19 In a recent study on 40 athletes studied after different endurance events (from marathon to ultra-endurance triathlon, 3–11 h in race duration), we reported an overall reduction in end-diastolic and end-systolic LV volumes immediately after the race, in sharp contrast to dilation of the RV (in both phases).22 Moreover, RV ejection fraction decreased immediately after the event (left ventricular ejection fraction – LVEF was unchanged), recovering to normal after 1 week. Intriguingly, the degree of transient RV dysfunction was significantly related to the duration of the endurance event. Moreover, BNP and cardiac troponin-I increases correlated with reductions in right ventricular ejection fraction (r = 0.52, p = 0.001 and r = 0.49, p = 0.002, respectively), but not LVEF.22 These findings support our 2003 hypothesis that endurance events lead to RV insults (reflected in enzyme rise and slight dysfunction), recovering after a single bout, but that repetitive ‘hits’ in the long-term may result in RV dysfunction and arrhythmias (figure 1). There may be a limit to what is healthy for the RV.

Healthy training versus overtraining of the heart. Healthy training with balanced exercise and recovery results in physiological remodelling in which enhanced cardiac structure and function enable greater cardiac performance during exercise. However, we propose that excessive exercise (training which is too intense and/or recovery that is too short) may cause cardiac injury and proarrhythmic remodelling which predominantly affects the right ventricle (adapted from the doctoral thesis of A La Gerche 2010).
We have performed detailed quantitative RV angiographic evaluation in 22 high-level endurance athletes presenting with ventricular arrhythmias, and compared those with age- and sex-matched comparable athletes (n = 15) and non-athletes (n = 10).23 Twenty-seven per cent of the athletes with arrhythmias had definite task force criteria for ARVC, although only 2/22 had a familial history. Four different software algorithms were used to measure RV and LV volumes and EF from biplane RV angiography. All athletes had normal LV function. Athletes with arrhythmias had a small but highly significantly lower RV ejection fraction than athletes without arrhythmias and controls (49.1% vs 63.7% vs 67%; p < 0.001).23 This decrease is clearly less pronounced than what is known from familial series of ARVC, but again points to underlying structural changes in the RV that may be associated with the observed arrhythmogenicity. Of note, many of these angiographies, when evaluated qualitatively by an experienced evaluator, were labelled as ‘normal for athlete's heart’, indicating the importance of quantitative evaluation as stated also in the revised ARVC task force criteria.24
What Defines Susceptibility To Exercise-Induced Rv Arrhythmogenicity?
As mentioned, 80% of the athletes whom we have evaluated with this syndrome were high-level (often competitive) cyclists or triathlon athletes.3 ,23 Cyclists perform the most protracted exercise, more hours per day and more days per year than any other athlete. They frequently sustain heart rates around 80% of maximum for prolonged periods. Their protracted aerobic exercise is regularly interrupted by intense anaerobic dashes, which is more uncommon in other endurance athletes: long-distance runners are notably scarce in our series, although this sport is very popular in our region. Therefore, the RV effects seem to be linked to high-intensity endurance activities, particularly those that are of long duration and combine endurance and power. We have observed the same presentation in rowers, triathletes and swimmers. We suspect that other sports with a similar physiological load, for example cross-country skiing, may also be associated with an increase in RV arrhythmias but this sporting demographic is not part of our experience.
However, not all such athletes develop RV dysfunction at the end of an event, and the incidence of long-term (acquired) RV cardiomyopathy is seemingly small. No formal estimates exist due to the lack of centralised registries. There is both uncertainty about the numerator (complete registry of cases?) and denominator (population of intense endurance athletes at risk?). Rough estimates based on the referral pattern for our tertiary care centre and the number or registered competitive and high-level recreational cyclists in our country, show that the yearly incidence for developing the phenotype may be around 1/1000 overall, but it may be 1/100 in international top-level athletes in the mentioned disciplines. Its relatively low prevalence is further highlighted by a normal survival curve in 119 former athletes participating to the Tour of Switzerland between 1975 and 1995: the observed survival was the same as that of a matched Swiss male population.25 Nevertheless, the question remains as to which individual facilitating factors may play a role in developing exercise-induced ARVC.
Illicit drug use?
The first suspect is always performance-enhancing drugs. Illicit drug use may be common in high-level endurance athletes although no reliable data exist on its prevalence. However, for most modern performance-enhancing drugs, no direct cardiopathic effect has been described.26 Moreover, if they were the direct cause, it is unclear why they would selectively have impact on the RV. Amphetamines for instance, are known to cause LV micro-infarcts and scarring, with secondary arrhythmias that often originate in the LV. Therefore, we do not consider illicit drugs as the direct cause. Their use may however be involved indirectly, that is, by allowing longer and more strenuous-endurance activity more frequently, facilitating development of the phenotype.
Genetic predisposition?
Another underlying factor could be genetic predisposition, since desmosomal mutations and alterations are known as the basis for familial ARVC. It is well established that exercise activity promotes the development of RV dysfunction and arrhythmogenicity in mutation carriers,27 ,28 and a similar relation has recently been described for left cardiomyopathies due to lamin A/C mutations.29 Therefore, it is obvious to suspect that the similar phenotypes found in high-level athletes as in familial ARVC is due to unmasking latent mutations. We systematically evaluated the five desmosomal genes for mutations (by sequencing) and larger genetic re-arrangements (by multiplex ligation-dependent probe amplification) in a cohort of 47 athletes, of whom 87% had definite or probable criteria for an ARVC phenotype.30 The proportion with desmosomal mutations was much lower than that described for familial ARVC (13% vs ∼50%).31–37 If RV arrhythmogenicity were the early expression of a latent underlying genetic (desmosomal) mutation, we would have expected at least a similar prevalence as in familial forms. Moreover, familial ARVC was only present rarely (in 2/47 athletes).
Some have argued that there might be a lower mutation rate in sporadic ARVC cases than in familial cohorts, and that this could explain the low-mutation rate in our cohort. However, most studies have recruited sporadic ‘index cases’ by including unrelated cases as they present to their institution. Because it is principally a familial disease, it is clear that a proportion of index cases have a positive family history (between 21% and 38%).32 ,35–37 When analysing the four largest studies which have included index cases with and without a positive family history, the mutation rate varies between 34% and 56% in index cases without a family history. In none of these studies, does the rate of mutation differ between those with and without a family history.
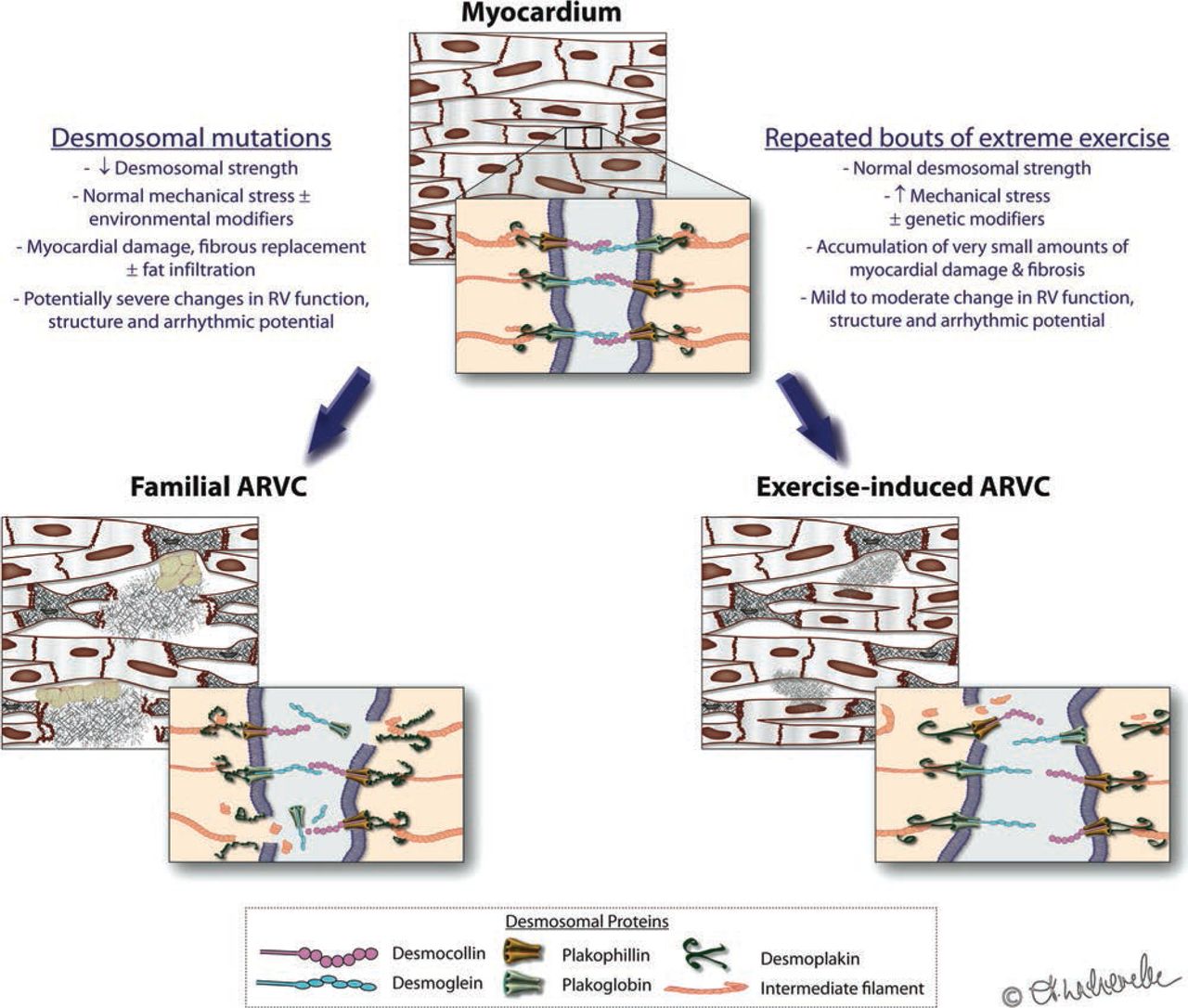
Although other genetic predisposition cannot be excluded, we do not believe that ‘exercise-induced ARVC’ is simply unmasking of mutation-dependent ‘familial ARVC’. Our genetic findings strengthen our initial hypothesis that excessive strain on the RV by sports itself can be regarded as one side of a continuous spectrum where myocardial integrity is perturbed due to a mismatch of strain and desmosomal integrity. With mutated desmosomes, normal levels of myocardial wall stress lead to cellular disruption and (fibro-fatty) repair, ending in familial ARVC. But we propose that a similar phenotype may develop when excessive wall stress disrupts ‘normal’ desmosomes, that is, in which the environmental factor plays the key role (figure 2). Like in other diseases, polygenetic factors, many outside the desmosomes, may interplay with environmental factors (here: exercise) to result in a phenotype. We do not believe however that cardiac monogenetic traits are or need to be the sole explanation for individual susceptibility.

Familial versus exercise-induced right ventricle (RV) cardiomyopathy: a continuum? Integrity of myocyte junctions is critical to cardiac function, structure and electrical stability. These junctions can be compromised by genetic mutations of the desmosomal proteins (left pane) or by increased mechanical stress (right pane). Either mechanism, or a contribution of both mechanical and genetic factors, may predispose to apoptosis, fibrosis and arrhythmogenicity, which most commonly affects the RV.
Moreover, it has been suggested that the molecular pathogenesis of ARVC has two components. The first component is fibroadiposis. The second is cardiac dysfunction, which is primarily due to impaired myocyte-to-myocyte attachment.38 The pathogenesis of fibroadiposis involves partial nuclear translocation of plakoglobin and subsequent suppression of canonical Wnt signalling, which is involved in the development of the RV and its outflow tract, the predominant sites of involvement in familial ARVC. This pathogenic pathway may not be at work in athletes, whereas the myocyte-to-myocyte disruption is common in both. This requires further study. The exact incidence and significance of subclinical (microscopic) fibrosis in athletes is difficult to quantify given that myocardial biopsies are seldom performed and those biopsies which have been performed tend to be in selected cohorts with more severe symptoms or changes in cardiac structure or function.3 ,30 ,39 An interesting recent exception to this is a study by Dello Russo et al40 in which athletes with ventricular arrhythmias but no overt structural heart disease were investigated with electroanatomical guided myocardial biopsy. There were histological abnormalities in all 13 of the athletes, which the authors attributed to ARVC, myocarditis or substance abuse. Whereas pathologic examination often shows fibrofatty replacement predominantly in the epicardial surface, probably because of increased wall stress on the outer surface, it is not known whether this is the case exercise induced ARVC and whether this is different from familial ARVC, since biopsies are taken endocardially and not transmurally. In our experience however, there is rarely endocardial voltage abatement in athletes with exercise-induced ARVC compared with those with familial ARVC forms. Data on epicardial mapping in athletes are lacking.
Haemodynamic stress during exercise: animal models
The fact that exercise alone, without doping or genetic predisposition, could lead to the phenotype of exercise-induced ARVC, finds further credence in animal models. There is no perfect model of endurance athletic activity but two models have merit. Earlier findings were reported in a model based on an induced aortacaval fistula in pigs,41 leading to volume and pressure overload of the RV, a disproportionate increase in RV stroke work index relative to that of the LV (+216% vs +70%), RV fibrosis and the development of RV dysfunction after 3 months. The pure LV volume overload was associated with hypertrophy based on myocyte length increase and increased local production of insulin-like growth factor. On the other hand, the RV showed more pronounced hypertrophy, increase in both myocyte length and diameter, and associated increased collagen deposition. Apart from IGF-I increase, there also was an increase in endothelin-I and angiotensin-II production. The authors postulated that the differential loads led to different gene expression and different structural changes.41
More recently, the Barcelona group of Lluis Mont et al have established a rat model of endurance activity.42 Although rats can be forced to run only for a maximum of 1 h/day (ie, much shorter than man), the running rats showed increased interstitial fibrosis in both atria and in the RV after 16 weeks, in contrast to none in the LV. This was associated with inducibility of ventricular arrhythmias in 42% of the exercise rats, versus only 6% in controls (p = 0.05). Cessation of exercise reversed the fibrotic changes. The question is whether fibrogenesis can also be prevented in man with much longer-standing exercise history. Moreover, it remains to be seen whether administration of an ACE-inhibitor or angiotensin receptor blocker could also lead to reversibility or prevention, offering further prospect for clinical medicine. No data, however, are available so far on possible preventive or reversible effects of these drugs in athletes with supraventricular or ventricular arrhythmias.
Recent studies have evaluated late gadolinium enhancement on cardiac magnetic resonance imaging (CMR) in arrhythmia-free endurance athletes after an endurance race, but did not find its presence even when cardiac enzymes were elevated postexercise.13 ,16 In our opinion, however, this does not exclude acute microstructural damage which is too small to be picked up by the limited resolution of the CMR examination. In contrast, two studies have shown areas of fibrosis in 13% and 50% of athletes with a long-standing history of endurance competition, again pointing to chronic remodelling and arrhythmogenicity.22 ,43 However, the majority of symptomatic athletes in our studies with exercise-induced ARVC did not have overt fibrosis nor adiposis on CMR and only a minority showed histological fibroadiposis.3 Hence, macrostructural fibroadiposis seems to be less a hallmark of exercise-induced ARVC than of familial ARVC.
Why The Rv?
This brings us to a third pathophysiological predisposing mechanism, which may lie outside the heart. Intriguing as it may be, why would the RV (selectively) undergo proarrhythmic structural changes in endurance athletes? Can we expect a mismatch between wall stress and desmosomal integrity in the RV during high-intensity sports (as postulated above), and is this more likely to occur in the RV than in the LV? Indeed, there are indications for this. We and others have observed that intense physical exercise is associated with an increase in pulmonary arterial pressures, which is much more pronounced in trained athletes than in volunteers.44 ,45 Studies using invasive pressure measures have demonstrated that although both ventricles have to realise the same cardiac output, the pulmonary vascular bed can only reduce its resistance by 30–50% during exercise as compared with greater reductions in systemic vascular resistance (usually in excess of 75%).46 ,47 We, and others, have attempted to characterise pulmonary vascular function using non-invasive measures, and have demonstrated a strong relationship between pulmonary artery pressure estimates and cardiac output.44 ,48 ,49 This relationship is steeper and more linear than the comparable pressure/output relationship in the systemic circulation. Furthermore, this relationship is similar in athletes and non-athletes, that is, training does not lead to adaptation, which results in reduced afterload for the RV during exercise.44 ,50 Thus, irrespective of athletic status, the harder the exercise, the greater the RV pressure demands and the larger the proportional difference between the demands placed on the RV and the LV. Given that athletes are trained to attain higher cardiac output than controls, they achieve significantly higher peak pulmonary pressures as compared with non-athletes (figure 3).

{kind=link}
{kind=link}
{kind=link}
Disproportionately greater right ventricle (RV) pressures and wall stress give rise to RV remodelling. (A) During exercise, there is a near linear increase in pulmonary artery pressures during exercise with cardiac output and this relationship is similar for athletes (brown) and non-athletes (green). In healthy subjects, greater exercise capacity predicts higher pulmonary artery pressures with well-trained athletes generating the highest pressures of all. (B) As compared with the left ventricular, relative increases in wall stress are greater for the RV during intense exercise, the result of which is healthy cardiac remodelling with a very slight RV dominance, which diminishes with de-training. Repeated bouts of excessive and prolonged RV wall stress may result in cumulative RV damage, which may predispose to arrhythmias. The degree to which this adverse remodelling may recover with de-training is unclear.
The higher pulmonary pressures during exercise lead to a greater RV load and stroke work during exercise than for the LV.44 ,45 Based on the Laplace equation, and including chamber size and wall thickness estimates from cardiac MR, we estimated the resulting wall stress in both ventricles. Compared with rest, the RV wall stress at peak exercise rises in athletes by 170% compared with rest, with only a 23% increase in the LV wall stress.50 Furthermore, we observed RV hypertrophy (both wall mass and dilation) which was proportionally greater than LV hypertrophy in athletes as compared with non-athletes, thus confirming a link between acute haemodynamic load and chronic ventricular remodelling.50 It also stands to reason that single bouts of extreme and prolonged haemodynamic stress may result in RV fatigue or damage, thus potentially explaining both the ‘leak’ of enzymes and transient RV dysfunction following endurance sports. As mentioned, we have found that the enzyme rise correlates with the degree of transient RV dysfunction, whereas no such correlation exists with LV measures.22 Myocardial inflammation, substrate deficiency and oxidative stress have all been proposed as potential mediators of postexercise cardiac injury.51 We contend that the primary cause is the mechanical and metabolic stress of the prolonged intense exercise and that this triggers cellular pathways of injury and/or repair in the RV more than the LV. The ‘marathon rat’ study of Benito, Mont et al42 supports this premise.
We described above that the pulmonary vasculature has similar properties in athletes and non-athletes. Nevertheless, based on transit of microbubbles, two distinct groups of pulmonary vascular behaviour can be distinguished.44 At rest, no athlete or control showed microbubble passage through the lungs. In contrast, in about half of the population, microbubbles pass the lung vasculature during exercise. Again, this proportion is similar in athletes and non-athletes, and thus it does not seem to be a ‘trainable’ characteristic. Interestingly, those athletes who show bubble transfer during exercise attain 16% higher peak cardiac outputs with lower peak pulmonary pressures and resistance, and an attenuated BNP rise. The physiological basis of this bubble transfer is still unclear (opening of shunts or distension of pulmonary capillaries?). But it is evident that such physiology results in less RV wall stress for a comparable workload. We may speculate that such a pulmonary vasculature is less prone to develop RV wall damage, but this hypothesis needs further study.
Perspectives And Clinical Relevance
This article definitely does not negate the clear fact that physical activity can reduce cardiovascular morbidity and mortality. Sport is good and essential for all. But we may have to realise that adaptation of the athlete's heart is not always able to accommodate the sustained haemodynamic loads placed upon it. In some, intense endurance activity can lead to cardiac sports injury in the form of supraventricular or ventricular arrhythmias. Although the number of data is increasing, the prevalence of life-threatening arrhythmic problems is definitely small. But like other sports injuries, recognising the problem in an early phase can prevent disaster and may lead to measures that allow safe and enjoyable continuation of sports participation.
Diagnosis of RV arrhythmogenicity is not straightforward. Even in athletes with documented arrhythmias, confirming RV damage and proving compromised prognosis (ie, differentiating it from benign idiopathic RV outflow tract extrasystoles) is cumbersome. It requires an extensive work-up with expert electrophysiological insight. The diagnosis mainly resides on electrical findings (including an invasive EP study in some) rather than imaging (with CRM normal in many patients). However, first-line physicians should develop an alertness when they hear suspicious symptoms (like exertional sudden dyspnoea or light headedness), see negative T-waves beyond V2 or record ventricular premature beats (VPB) with a left bundle branch morphology on ECG, note ≥2500 VPB on a 24 h Holter, or see exercise-induced arrhythmias during an exercise test.
Modern training regimens include high-altitude training, real or simulated in hypobaric oxygen chambers. This may result in improvements in endurance performance but we wonder whether this may also lead to an increase in the potential for cardiac ‘over-training’, particularly of the RV. It is well known that subjects living in high altitude (more than 3000 m) have higher resting pulmonary pressures and that these pressures increase to even significantly higher levels during exercise (29 mm Hg to 60 mm Hg in high-altitude subjects vs 12 mm Hg to 18 mm Hg in sea level subjects).52 If our hypothesis is true that increases in arterial pulmonary pressures put a strain on the RV, high-altitude training will exacerbate this.15 Modern training regimens therefore may promote RV dysfunction, which therefore should be closely monitored and better studied. Moreover, if our hypothesis is correct, the athletic community in general should reflect on the appropriateness of ever increasing demands on athletes. Young cyclists undertake gruelling training and competition schedules, often biking more than 200 days per year for more than 5 h. Recreational athletes engage in ever longer and harder activities. There is a social aura of competitiveness and excess performance during sports, rather than to highlight the important social and healthy merits of moderate physical activity.
Most of the data on RV function in athletes are measured at rest, or derived indirectly from echocardiography during exercise (like pressure estimates). Although good correlations between these echocardiographic measures and invasive pressure measurements have been shown, direct and combined pressure-volume measurements during exercise are desirable.53 Such studies, combining RV imaging during exercise and invasive pressure measurements, are now underway. They may shed more light on this intriguing pathophysiology and help us to define risk patterns. Early recognition of those at risk would not only help prevent accidents, but also mostly help to reassure many others that they can enjoy their athletic endeavours safely.
Key Points
High-level endurance exercise is associated with high cardiac output, which results in high-pulmonary pressures and high right ventricular wall stress.
This high RV wall stress may explain findings of troponin rise and transient RV dysfunction after endurance sports events, due to putative cell rupture.
It is postulated that repetitive RV microtrauma may result in chronic RV structural and functional changes, and proarrhythmogenicity, in some endurance athletes. This syndrome is called ‘exercise-induced RV arrhythmogenic cardiomyopathy’.
Since desmosomal mutations are very uncommon in such athletes, we postulate that there might be a continuum between familial ARVC (with a mismatch between RV strain and desmosomal integrity due to weakened desmosomes) and exercise-induced ARVC (when excessive wall stress disrupts normal desmosomes).
Contributors All authors have contributed to the manuscript.
Competing interests HH is holder of the AstraZeneca Chair in Cardiac Electrophysiology, University of Leuven. HH received research funding through the University of Leuven from Siemens Medical Solutions. HH is Coordinating Clinical Investigator for the Biotronik-sponsored EuroEco study on health-economics of remote device monitoring. HH is a member of the scientific advisory board of Biosense Webster, St Jude Medical, Siemens Medical Solutions, Boehringer-Ingelheim, Bayer and Sanofi-Aventis, and receives unconditional research grants through the University of Leuven from St Jude Medical, Medtronic, Biotronik and Boston Scientific Inc. DP is member of scientific advisory boards for Actelion and Servier. ALG receives a post-doctoral research scholarship from the Australian National Health and Medical Research Council.
Provenance and peer review Commissioned; externally peer reviewed.