
Abstract
Aims/hypothesis
The aim of this study was to determine whether adiponectin elicits glucose uptake via increased GLUT4 translocation and to investigate the metabolic fate of glucose in skeletal muscle cells treated with globular adiponectin.
Materials and methods
Basal and insulin-stimulated 2-deoxy-d-[3H]glucose uptake, cell surface myc-tagged GLUT4 content, production of 14CO2 by oxidation of d-[U-14C]glucose and [1-14C]oleate, and incorporation of d-[U-14C]glucose into glycogen and lactate were measured in the presence and absence of globular adiponectin.
Results
RT-PCR and Western blot analysis revealed that L6 cells and rat skeletal muscle cells express AdipoR1 mRNA and protein. Globular adiponectin increased both GLUT4 translocation and glucose uptake by increasing the transport V max of glucose without altering the K m. Interestingly, the incorporation of d-[U-14C]glucose into glycogen under basal and insulin-stimulated conditions was significantly decreased by globular adiponectin, whereas lactate production was increased. Furthermore, globular adiponectin did not affect glucose oxidation, but enhanced phosphorylation of AMP kinase and acetyl-CoA carboxylase, and fatty acid oxidation.
Conclusions/interpretation
The present study is the first to show that globular adiponectin increases glucose uptake in skeletal muscle cells via GLUT4 translocation and subsequently reduces the rate of glycogen synthesis and shifts glucose metabolism toward lactate production. These effects are consistent with the increased phosphorylation of AMP kinase and acetyl-CoA carboxylase and oxidation of fatty acids induced by globular adiponectin.
Similar content being viewed by others


Introduction
Adiponectin (Acrp30) [1] is a 30-M r adipose-derived factor that can directly affect glucose homeostasis and regulate peripheral insulin sensitivity [2–5]. Injection of adiponectin into rodents has been shown to decrease resting blood glucose levels and protect animals with diet-induced obesity from developing insulin resistance [6–9]. Treatment of isolated skeletal muscle or C2C12 myocytes with adiponectin stimulates glucose transport [6, 10, 11], suggesting that this may be one mechanism by which adiponectin acts to alleviate hyperglycaemia. However, very little is known about the molecular mechanism by which adiponectin stimulates glucose uptake.
Physiological stimuli, such as insulin and exercise, stimulate glucose uptake in skeletal muscle by mobilising GLUT4 to the plasma membrane [12]. These stimuli are known to utilise different signalling pathways to elicit glucose uptake. Insulin stimulates GLUT4 translocation via a phosphatidylinositol 3-kinase-dependent signalling pathway [13, 14], whereas AMP kinase plays an important role in mediating the effect of exercise [15]. Insulin acts to divert a large proportion of the glucose that enters muscle towards glycogen synthesis. In fact, it has been reported that the synthesis of muscle glycogen accounts for the majority of whole-body glucose uptake and virtually all non-oxidative glucose metabolism in both normal and diabetic subjects [16]. Interestingly, the activation of AMP kinase increases glucose uptake in muscle cells and switches on catabolic pathways that generate ATP (e.g. fatty acid oxidation) while switching off cellular processes that consume ATP, specifically, biosynthetic pathways (e.g. glycogen synthesis) [15]. Therefore, if the function of AMP kinase is to monitor cellular energy status and adiponectin activates this kinase, one would expect the anabolic processes in the cell to be suppressed following adiponectin treatment. This would lead glucose to a metabolic fate distinct from that determined by insulin in the muscle cell.
Although it has been reported that adiponectin increases glucose uptake in skeletal muscle, apparently via activation of AMP kinase [6], it remains to be determined whether adiponectin increases GLUT4 translocation in skeletal muscle. Additionally, very little is currently known about the metabolic fate of the extra glucose that enters the muscle cells after adiponectin treatment. In the present study we investigated the effects of globular adiponectin (gAd) on GLUT4 translocation, glucose uptake, glycogen synthesis, glucose oxidation, lactate production and fatty acid oxidation in muscle cells in culture. We also determined the effect of adiponectin on the phosphorylation of AMP kinase and acetyl-CoA carboxylase (ACC).
Materials and methods
Chemicals
We purchased 5-amino-4-imidazole carboxamide riboside (AICAR) from Toronto Research Chemicals (Toronto, ON, Canada). Cytochalasin B, glycogen, O-phenylenediamine dihydrochloride (OPD), defatted BSA and phenylethylamine were purchased from Sigma (St. Louis, MO, USA). Human insulin (Humulin R) was obtained from Eli Lilly (Toronto, ON, Canada), and recombinant human gAd produced in Escherichia coli was obtained from Peprotech (Ottawa, ON, Canada). We purchased d-[U-14C]glucose, [1-14C]oleate and 2-deoxy-d-[3H]glucose from Amersham (Baie d’Urfe, QC, Canada). Cell culture components were supplied by Wisent (St. Foy, QC, Canada). All other chemicals used were of the highest grade available.
Cell culture conditions for L6 myotubes
For all experiments, L6 rat skeletal muscle cells were routinely grown in minimum essential medium alpha (α-MEM) supplemented with 10% (v/v) fetal bovine serum in a humidified atmosphere of 95% air and 5% CO2 at 37°C. For differentiation into myotubes, the cells were grown in medium containing 2% fetal bovine serum for 5–7 days. We employed cells transfected to stably overexpress GLUT4 harbouring a myc epitope on the first exofacial loop of the transporter (L6 GLUT4myc cells; a kind gift from A. Klip, The Hospital for Sick Children, Toronto, ON, Canada). This facilitated accurate quantitative analysis of GLUT4 translocation in intact cells [14, 17, 18].
Analysis of adiponectin receptor-1 expression by RT-PCR and western blotting
Primers based on the putative nucleotide sequence for rat adiponectin receptor-1 (AdipoR1; a kind gift from M. Wheeler, University of Toronto) were used to investigate the expression of this receptor in L6 rat skeletal muscle cells. RNA was isolated from rat soleus muscle cells and L6 rat skeletal muscle cells using the Trizol reagent from Invitrogen (Burlington, ON, Canada). Reverse transcription was performed using the Enhanced Avian Reverse Transcriptase Kit from Sigma with 1 μg of total RNA and 1 U of reverse transcriptase according to the manufacturer’s recommendations. The sequences of the forward and reverse primers were 5′-GGAAGATCTGCCAGATGTCTTCCCACAAAG-3′ and 5′-CGCGGATCCGAGGCTCAGAGAAGGGAGTCG-3′, respectively. Using these primers the length of the PCR product for AdipoR1 should be 1,128 bp. AdipoR1 cDNA was amplified for 35 cycles in a 50-μl reaction containing 1×PCR buffer (50 mmol/l KCl, 10 mmol/l Tris–HCl, pH 8.3), 200 μmol/l of each dNTP, 1.5 mmol/l MgCl2, 0.1 μmol/l forward and reverse primers and 1 U of JumpStart AccuTaq Polymerase (Sigma). The thermal cycling program was set to 94°C for 30 s, following a hybridisation step at 60°C for 30 s, and an elongation step at 72°C for 90 s. The resulting cDNA was evaluated by electrophoresis through a 1% agarose gel followed by ethidium bromide staining. Total membranes were prepared from L6 cells as previously described [19] and then analysed by Western blotting for the presence of AdipoR1. Western blotting was conducted as described below, in this case using a primary antibody (1:750 dilution; Alpha Diagnostic International, San Antonio, TX, USA) that specifically recognised AdipoR1 and a goat anti-rabbit secondary antibody (1:10,000 dilution; Cell Signaling Beverley, MA, USA).
Measurement of glucose uptake
L6 myotubes were cultivated in 12-well plates and serum-starved for 4 h before being incubated with or without gAd (6–240 μmol/l, 2 h) and/or insulin (100 nmol/l, 20 min) or AICAR (1 mmol/l, 30 min). Myotubes were subsequently washed twice, and glucose transport was assayed in HEPES–buffered saline solution (140 mmol/l NaCl, 20 mmol/l HEPES–Na, 2.5 mmol/l MgSO4, 1 mmol/l CaCl2, 5 mmol/l KCl, pH 7.4) containing 10 μmol/l 2-deoxyglucose (18,500 Bq/ml 2-deoxy-d-[3H]glucose) as described previously [17]. For kinetic measurements, cells were grown and differentiated in 96-well plates and volumes were adjusted accordingly. Glucose uptake was determined in HEPES–buffered saline solution containing 0.005–10 mmol/l 2-deoxyglucose (185 kBq/ml 2-deoxy-d-[3H]glucose). The incubation medium was aspirated, then cells were washed with ice-cold saline and lysed with 1 ml of NaOH (0.05 mol/l). Cell lysates were transferred to scintillation vials for radioactivity counting. Non-specific uptake was determined in the presence of cytochalasin B (10 μmol/l) and was subtracted from all values. Apparent K m and V max values were calculated by the method of Eadie–Hoftsee as explained previously [20]. This analysis assumes that 2-deoxglucose transport displays simple hyperbolic kinetics.
Determination of cell surface GLUT4myc
The exofacial location of the myc epitope on GLUT4 in L6 GLUT4myc cells allows the analysis of cell surface GLUT4 in intact cells. Levels of GLUT4myc at the cell surface were measured by an antibody-coupled colorimetric assay as described previously [17, 21]. Briefly, L6 myotubes were cultivated in 24-well plates and serum-starved for 4 h before being incubated with or without gAd (60 μmol/l, equivalent to 1 μg/ml, 2 h) and/or insulin (100 nmol/l, 20 min) or AICAR (1 mmol/l, 30 min). Cells were then quickly washed in ice-cold PBS and incubated with an anti-c-myc antibody (1:200 dilution; A14; Santa Cruz Biotech, Santa Cruz, CA, USA) at 4°C for 60 min. Cells were washed and fixed in 3% paraformaldehyde for 3 min on ice. The fixative was neutralised by incubation in 10 mmol/l glycine in ice-cold PBS for 10 min. Cells were blocked in 10% goat serum and 3% BSA for 30 min, and then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:1,000 dilution; Cell Signaling) at 4°C for 60 min. Cells were washed five times with ice-cold PBS and then incubated with OPD reagent (1 ml/well) at room temperature for 30 min. The reaction was stopped by the addition of 0.25 ml of 3 mol/l HCl. The supernatant was collected and its absorbance at 492 nm was measured.
Measurement of lactate production
L6 myotubes were starved of serum for 4 h and then incubated with or without gAd (60 μmol/l) and/or insulin (100 nmol/l) for 2 h. Following the incubation period, the media were collected for lactate determination. The total lactate released in the medium was measured as described previously [21] using the lactate oxidase assay kit from Sigma Diagnostics (St. Louis, MO, USA).
Measurement of glycogen synthesis
Glycogen synthesis was assessed by measuring the incorporation of d-[U-14C]glucose into glycogen as described previously [21]. Briefly, following 4 h of serum starvation, L6 myotubes in 6-well plates were incubated for 2 h in medium containing 0.15 μCi/ml of d-[U-14C]glucose with or without gAd (60 μmol/l) and/or insulin (100 nmol/l). Cells were then quickly washed in ice-cold PBS and lysed in 0.2 ml of KOH (1 mol/l). Cell lysates were used for overnight glycogen precipitation with ethanol. Precipitated glycogen was dissolved in water and transferred to scintillation vials for radioactivity counting. An aliquot of the cell lysates was used for protein determination.
Conversion of d-[U-14C]glucose and [1-14C]oleate into 14CO2
Glucose and oleate oxidation were assessed by measuring the production of 14CO2 from d-[U-14C]glucose and [1-14C]oleate as described previously [21] with slight modifications. Briefly, L6 myotubes were incubated for 2 h with serum-free medium containing either 5,550 Bq/ml of d-[U-14C]glucose or 11 kBq/ml [1-14C]oleate with or without insulin (100 nmol/l) and/or gAd (60 μmol/l) and/or AICAR (1 mmol/l) in 60×15-mm petri dishes. The [1-14C]oleate and cold oleate (0.5 mmol/l final concentration) were complexed with defatted BSA at a ratio of 2.5:1. A piece of Whatman paper wet with 100 μl of phenylethylamine/methanol (1:1) was taped onto the inside of each petri dish to trap the CO2 produced during the incubation period, and parafilm was used to seal the dishes. After a 2-h incubation period, 200 μl of H2SO4 (4 mol/l) was added to the cells, which were then further incubated for 1 h at 37°C. Finally, the seal was removed from each petri dish and the pieces of Whatman paper were carefully transferred to scintillation vials for radioactivity counting.
Western blot determination of AMP kinase and ACC phosphorylation
L6 myotubes were grown in 6-well plates and serum-starved for 4 h. Following treatment with or without gAd (60 μmol/l, 30 min) and/or AICAR (1 mmol/l, 30 min), cells were washed twice in ice-cold PBS. Treatment with AICAR was used as a positive control for AMP kinase and ACC phosphorylation. Cells were subsequently lysed in buffer containing 135 mmol/l NaCl, 1 mmol/l MgCl2, 2.7 mmol/l KCl, 20 mmol/l Tris (pH 8.0), Triton 1%, glycerol 10%, and protease and phosphatase inhibitors (0.5 mmol/l Na3VO4, 10 mmol/l NaF, 1 μmol/l leupeptin, 1 μmol/l pepstatin, 1 μmol/l okadaic acid, 0.2 mmol/l PMSF), passed through a 25-gauge syringe five times, and then heated (65°C, 5 min). An aliquot of the cell lysates was used to determine the protein concentration in each sample. Prior to loading onto SDS-PAGE gels, the samples were diluted 1:1 (v/v) with 2× Laemmli sample buffer. The buffer consisted of 62.5 mmol/l Tris–HCl (pH 6.8), 2% (w/v) SDS, 50 mmol/l DTT, 0.01% (w/v) bromophenol blue. Aliquots of cell lysates containing 25 μg of protein were then subjected to SDS-PAGE (8–10% resolving gels), and then transferred to polyvinylidene difluoride (PVDF) membranes (Bio-Rad, Burlington, ON, Canada). The phosphorylation of AMP kinase was determined using a phospho-AMP kinase (Thr-172)-specific antibody (1:1,000 dilution; Cell Signaling), and ACC phosphorylation was detected using a phospho-ACC-specific antibody (1:500 dilution; Upstate Biochemicals, Charlottesville, VA, USA) that recognises Ser-79 of ACC1 and Ser-227 of ACC2.
Statistical analysis
Statistical analyses were performed by one-way ANOVA with either Tukey–Kramer or Bonferroni post hoc tests. A p value less than 0.05 was considered significant.
Results
Adiponectin receptor in L6 muscle cells
RT-PCR analysis showed a product matching the expected size of 1,128 bp, indicating that AdipoR1 mRNA is present in both rat soleus muscle cells and L6 rat skeletal muscle cells (Fig. 1a). Expression of the AdipoR1 protein was confirmed by Western blotting total membrane samples from L6 cells with an AdipoR1-specific antibody (Fig. 1b).

a RT-PCR of AdipoR1 expression. Lane I shows the DNA ladder. A product matching the expected size of 1,128 bp indicates that AdipoR1 mRNA is present in both rat soleus muscle cells (Lane II) and L6 rat skeletal muscle cells (Lane III). b Western blot analysis. An antibody that specifically recognises AdipoR1 was used to confirm the presence of AdipoR1 (∼42 M r) in total membranes prepared from L6 cells
Glucose uptake and cell surface GLUT4myc content
The effect of a range of gAd concentrations on glucose uptake was tested; a significant increase in basal and insulin-stimulated glucose uptake was elicited by gAd at concentrations of 30 μmol/l and higher (Fig. 2). Importantly, as shown in Table 1, gAd increased glucose uptake by altering the V max and had no effect on the K m. AICAR is used experimentally to stimulate AMP kinase as it readily enters cells and is subsequently phosphorylated to the monophosphate form (ZMP) which mimics the effect of AMP on the AMP kinase cascade [22]. AICAR increased glucose uptake (∼1.8-fold), but the combination of gAd and AICAR did not cause any increase in glucose uptake above their respective individual effects (Fig. 3a). In contrast, when used in combination with insulin, gAd exerted an additive effect, increasing the magnitude of insulin-stimulated glucose uptake by ∼27% (Fig. 3a). Analysis of the GLUT4myc content in the plasma membrane produced similar results to those obtained for glucose uptake. The GLUT4myc content was significantly increased by insulin (∼2.3-fold), gAd (∼1.4-fold) and AICAR (∼1.6-fold), and gAd had an additive effect in combination with insulin, increasing the cell surface GLUT4myc content by ∼22% above that observed on stimulation with insulin alone (Fig. 3b).

Basal and insulin-stimulated glucose uptake response of L6 myotubes to increasing concentrations (0, 6, 30, 60, 120 and 240 μmol/l) of gAd. The insulin-stimulated myotubes were exposed to 100 nmol/l of insulin for 20 min prior to the glucose uptake assay. The data are representative of three independent experiments and are expressed as means±SEM; Filled-circles:, +insulin; open circles: basal. *p<0.05 vs control under basal conditions; #p<0.05 vs insulin-stimulated control

Effect of gAd (60 μmol/l, equivalent to 1 μg/ml, 2 h), insulin (100 nmol/l, 20 min) and AICAR (1 mmol/l, 30 min), either alone or in combination, on 2-deoxyglucose uptake (a) and plasma membrane GLUT4myc content (b) in L6 GLUT4myc myotubes. The data are representative of four independent experiments and are expressed as means±SEM. *p<0.05 vs control under basal conditions; #p<0.05 vs insulin-stimulated control. Con control, Ins insulin
Glycogen synthesis and lactate production
The incorporation of d-[U-14C]glucose into glycogen was significantly increased in the presence of insulin (∼2-fold). Interestingly, gAd significantly reduced both the basal and the insulin-stimulated rates of d-[U-14C]glucose incorporation into glycogen by ∼28 and 60%, respectively (Fig. 4a). The production of lactate was significantly increased by insulin (∼30%) and gAd (∼40%) (Fig. 4b). Furthermore, gAd exerted a significant additive effect, increasing the insulin-stimulated rate of lactate production in L6 myotubes by ∼48% (Fig. 4b).

Effect of gAd (60 μmol/l, 2 h) and insulin (100 nmol/l, 2 h), either alone or in combination, on the incorporation of d-[U-14C]glucose into glycogen (a) and lactate production (b) in L6 rat skeletal muscle cells. The data are representative of four independent experiments and are expressed as means±SEM. *p<0.05 vs control; #p<0.05 vs control, and Ins and gAd alone. Con control, Ins insulin
Oxidation of glucose and oleate
In the presence of insulin, the production of 14CO2 from d-[U-14C]glucose was significantly increased by ∼35% (Fig. 5). Incubation of L6 myotubes with AICAR and gAd did not affect the basal or insulin-stimulated rates of 14CO2 production from d-[U-14C]glucose (Fig. 5), whereas both significantly increased (∼1.7-fold) the basal rates of 14CO2 production from [1-14C]oleate (Fig. 6). The combination of AICAR and gAd did not have an additive effect on the production of 14CO2 from [1-14C]oleate (Fig. 6).

Effect of gAd (60 μmol/l, 2 h), insulin (100 nmol/l, 2 h), AICAR (1 mmol/l, 2 h) and gAd plus insulin on 14CO2 production from d-[U-14C]glucose. The data are representative of four independent experiments and are expressed as means±SEM. *p<0.05 vs control. Con control, Ins insulin

Effect of gAd (60 μmol/l, 2 h) and AICAR (1 mmol/l, 2 h), either alone or in combination, on 14CO2 production from [1-14C]oleate in L6 rat skeletal muscle cells. The data are representative of four independent experiments and are expressed as means±SEM. *p<0.05 vs control. Con control
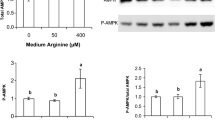
AMP kinase and ACC phosphorylation
In the presence of gAd or AICAR, the phosphorylation of AMP kinase was significantly increased by ∼2-fold; the combination of gAd and AICAR did not cause any additional effects (Fig. 7a). Both gAd and AICAR also caused a significant increase (∼2-fold) in the phosphorylation of ACC (Fig. 7b), a well-characterised substrate of AMP kinase, confirming that phosphorylation of AMP kinase led to the activation of its kinase activity.

Effect of gAd (60 μmol/l, 30 min) and AICAR (1 mmol/l, 30 min), either alone or in combination, on AMP kinase and ACC phosphorylation in L6 rat skeletal muscle cells. Representative blots of phosphorylated AMP kinase (a) and phosphorylated ACC (b) are shown in the panels above the graphs showing data from two independent quantitative analysis experiments. The data are expressed as means±SEM. *p<0.05 vs control. Con control
Discussion
Adiponectin circulates in plasma in various forms, including trimeric, hexameric and higher order complexes [23]. A cleaved fragment containing the globular head group of adiponectin (gAd) has also been found circulating in human plasma [9], and the physiological importance of this fragment has been highlighted by recent studies reporting the correction of diabetes and insulin resistance in transgenic ob/ob mice expressing gAd [7]. Each of the circulating forms of adiponectin appears to mediate distinct, often tissue-specific, metabolic effects. The recent identification of two receptors for adiponectin [6], AdipoR1 and AdipoR2, has provided a possible mechanism by which cleavage of the full-length protein to liberate the globular domain may control the physiological function of adiponectin. Of the two receptors, AdipoR1, which is predominantly expressed in skeletal muscle, exhibits a higher affinity for gAd [6]. Indeed, injection of recombinant protein representing the globular domain has been demonstrated to cause a rapid increase in muscle NEFA oxidation and to prevent diet-induced obesity without an alteration in food intake [9]. However, in contrast to muscle, liver cells predominantly express AdipoR2 and appear to respond only to the full-length protein [6]. In the present study we used two approaches to demonstrate that L6 rat skeletal muscle cells express AdipoR1 mRNA and protein. We subsequently investigated the effects of gAd on glucose uptake and metabolism in these cells.
Consistent with previous observations in rat skeletal muscle and C2C12 myocytes [6, 10, 11], gAd increased glucose uptake in L6 myotubes. Importantly, we have demonstrated that gAd increases glucose uptake by increasing the cell surface content of GLUT4myc. To our knowledge, this is the first time that adiponectin has been shown to elicit GLUT4 translocation. This observation is in line with our data indicating that gAd increases the transport V max for glucose without altering the K m. Previous studies have suggested that adiponectin regulates glucose uptake via an AMP kinase-dependent mechanism [6, 10]. AMP kinase activation by AICAR has been shown to increase glucose uptake via [24, 25], or independently of [26] GLUT translocation. In our studies, the fact that gAd had an additive effect on insulin-stimulated glucose uptake suggests that they may act via different mechanisms. Furthermore, the combination of gAd and AICAR did not have an additive effect on glucose uptake, GLUT4myc translocation, or AMP kinase or ACC phosphorylation, suggesting that gAd increases GLUT4 translocation and glucose uptake via an insulin-independent mechanism involving AMP kinase.
Previous studies have reported that adiponectin increases glucose uptake in muscle cells [6, 10, 11] and fat cells [27], but few have examined the subsequent metabolism of glucose. Potential pathways of glucose metabolism in muscle cells include glycogen synthesis, glucose oxidation and lactate production [28]. In the present study, adiponectin increased glucose uptake, but reduced the basal and insulin-stimulated rates of glycogen synthesis in muscle cells. Given that a previous study reported that full-length adiponectin had no effect on whole-body glucose uptake or glycogen synthesis [29], this indicates that gAd specifically mediates these processes via AdipoR1 in skeletal muscle. The observed reduction in glycogen synthesis was accompanied by a proportional increase in lactate production. A study using C2C12 cells also recently reported a small increase in lactate production in response to gAd [6]. The increased glycolytic flux stimulated by adiponectin did not alter the oxidation of glucose, suggesting that the production of lactate became the main metabolic pathway by which glucose was processed in adiponectin-treated muscle. Typically, lactate produced by skeletal muscle promptly diffuses into the circulation and may be used as a substrate for glycogen synthesis in the liver or for oxidation in the heart. However, to date, no studies have examined the effect of adiponectin treatment on plasma lactate levels.
The signalling pathways regulated by adiponectin are only just beginning to be characterised. Current evidence suggests that many effects of this molecule are mediated via AMP kinase and that this kinase plays a central role in mediating the metabolic effects of adiponectin [6, 30]. AMP kinase is a multisubstrate heterotrimer that contains a catalytic α-subunit and two regulatory (β and γ) subunits which play an important role in determining substrate specificity and protein stability [15]. AMP kinase is allosterically activated by increases in the ratio of AMP : ATP or creatine : phosphocreatine, and by phosphorylation on Thr-172. It regulates a series of responses that act to restore energy balance in cells, switching off ATP-consuming pathways and switching on ATP-generating pathways which, as shown here, include increased glucose uptake and fatty acid oxidation in muscle. Recent studies have identified an interaction between AMP kinase activity and glycogen content in cells. It has been reported that AMP kinase can directly phosphorylate and inactivate glycogen synthase in vitro [31]. It has also been demonstrated that the β-subunit of AMP kinase contains a motif that can bind glycogen [32, 33], and increased glycogen binding is correlated with decreased AMP kinase activation in response to AICAR [34]. These data suggest that AMP kinase activation promotes depletion of cellular glycogen stores, and this is consistent with our results showing that adiponectin activates AMP kinase and reduces glycogen synthesis in L6 rat skeletal muscle cells.
The accumulation of lipid in muscle and other tissues has been linked to the development of insulin resistance [35]. Previous work has shown that adiponectin increases fat oxidation in muscle and liver, and that AMP kinase appears to play a central role in the regulation of lipid metabolism by adiponectin [6, 9, 10]. The transport of fat into mitochondria via the carnitine palmitoyl transferase 1 (CPT-1) transporter is required for oxidation to occur and this process is inhibited by malonyl CoA [10, 36]. It has been suggested that the insulin-sensitising effects of adiponectin may be due to a decrease in malonyl CoA levels and increased fat oxidation [36]. In the present study we have demonstrated that adiponectin increases fatty acid oxidation in L6 cells, and this is likely to be associated with enhanced insulin sensitivity. We have also shown that gAd increases the phosphorylation of, and thus inactivates ACC. This would be expected to decrease malonyl CoA levels, thereby reversing the inhibition of CPT-1 and increasing fat transport into mitochondria [37]. Indeed, the increased phosphorylation of ACC was observed to coincide with an increased rate of fatty acid oxidation in our study.
In summary, we have shown that gAd increases glucose uptake, GLUT4myc translocation, fatty acid oxidation and lactate production, and decreases basal and insulin-stimulated rates of glycogen synthesis. These metabolic effects observed are consistent with the phosphorylation and activation of AMP kinase and the inhibition of ACC. This study is the first to address the changes that occur in glucose metabolism following the gAd-stimulated increase in glucose uptake and fatty acid metabolism in L6 rat skeletal muscle cells.
Abbreviations
- ACC:
-
acetyl-CoA carboxylase
- CPT-1:
-
carnitine parmitoyl transferase 1
- gAd:
-
globular adiponectin
- AdipoR:
-
adiponectin receptor
- AICAR:
-
5-amino-4-imidazole carboxamide riboside
References
Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF (1995) A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem 270:26746–26749
Berg AH, Combs TP, Scherer PE (2002) ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab 13:84–89
Pajvani UB, Scherer PE (2003) Adiponectin: systemic contributor to insulin sensitivity. Curr Diabetes Rep 3:207–213
Wolf G (2003) Adiponectin: a regulator of energy homeostasis. Nutr Rev 61:290–292
Tsao TS, Lodish HF, Fruebis J (2002) ACRP30, a new hormone controlling fat and glucose metabolism. Eur J Pharmacol 440:213–221
Yamauchi T, Kamon J, Ito Y et al (2003) Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 423:762–769
Yamauchi T, Kamon J, Waki H et al (2003) Globular adiponectin protected ob/ob mice from diabetes and apoE-deficient mice from atherosclerosis. J Biol Chem 278:2461–2468
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001) The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:947–953
Fruebis J, Tsao TS, Javorschi S et al (2001) Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A 98:2005–2010
Tomas E, Tsao TS, Saha AK et al (2002) Enhanced muscle fat oxidation and glucose transport by ACRP30 globular domain: acetyl-CoA carboxylase inhibition and AMP-activated protein kinase activation. Proc Natl Acad Sci U S A 99:16309–16313
Maeda N, Shimomura I, Kishida K et al (2002) Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 8:731–737
Tremblay F, Dubois MJ, Marette A (2003) Regulation of GLUT4 traffic and function by insulin and contraction in skeletal muscle. Front Biosci 8:d1072–d1084
Somwar R, Niu W, Kim DY et al (2001) Differential effects of phosphatidylinositol 3-kinase inhibition on intracellular signals regulating GLUT4 translocation and glucose transport. J Biol Chem 276:46079–46087
Sweeney G, Garg R, Ceddia R et al (2004) Intracellular delivery of phosphatidylinositol (3,4,5)-trisphosphate causes translocation and incorporation of GLUT4 into the plasma membrane of muscle and fat cells without increasing transport. J Biol Chem 279:32233–32242
Hardie DG (2004) AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Med Sci Sports Exerc 36:28–34
Shulman GI (2000) Cellular mechanisms of insulin resistance. J Clin Invest 106:171–176
Tajmir P, Kwan JJ, Kessas M, Mozammel S, Sweeney G (2003) Acute and chronic leptin treatment mediate contrasting effects on signaling, glucose uptake, and GLUT4 translocation in L6-GLUT4myc myotubes. J Cell Physiol 197:122–130
Sweeney G, Somwar R, Ramlal T, Volchuk A, Ueyama A, Klip A (1999) An inhibitor of p38 mitogen-activated protein kinase prevents insulin-stimulated glucose transport but not glucose transporter translocation in 3T3-L1 adipocytes and L6 myotubes. J Biol Chem 274:10071–10078
Sumitani S, Ramlal T, Liu Z, Klip A (1995) Expression of syntaxin 4 in rat skeletal muscle and rat skeletal muscle cells in culture. Biochem Biophys Res Commun 213:462–468
Somwar R, Kim DY, Sweeney G et al (2001) GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem J 359:639–649
Ceddia R, Sweeney G (2004) Creatine increases glucose oxidation and AMPK phosphorylation and reduces lactate production in L6 rat skeletal muscle cells. J Physiol 555:409–421
Corton JM, Gillespie JG, Hawley SA, Hardie DG (1995) 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 229:558–565
Waki H, Yamauchi T, Kamon J et al (2003) Impaired multimerization of human adiponectin mutants associated with diabetes. Molecular structure and multimer formation of adiponectin. J Biol Chem 278:40352–40363
Russell RR III, Bergeron R, Shulman GI, Young LH (1999) Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol 277:H643–H649
Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW (1999) 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48:1667–1671
Abbud W, Habinowski S, Zhang JZ et al (2000) Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of Glut1-mediated glucose transport. Arch Biochem Biophys 380:347–352
Wu X, Motoshima H, Mahadev K, Stalker TJ, Scalia R, Goldstein BJ (2003) Involvement of AMP-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes 52:1355–1363
Kelley DE, Mandarino LJ (2000) Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 49:677–683
Combs TP, Berg AH, Obici S, Scherer PE, Rossetti L (2001) Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J Clin Invest 108:1875–1881
Tsao TS, Tomas E, Murrey HE et al (2003) Role of disulfide bonds in Acrp30/adiponectin structure and signaling specificity: different oligomers activate different signal transduction pathways. J Biol Chem 278 50810–50817
Carling D, Hardie DG (1989) The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim Biophys Acta 1012:81–86
Hudson ER, Pan DA, James J et al (2003) A novel domain in AMP-activated protein kinase causes glycogen storage bodies similar to those seen in hereditary cardiac arrhythmias. Curr Biol 13:861–866
Polekhina G, Gupta A, Michell BJ et al (2003) AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol 13:867–871
Wojtaszewski JF, Jorgensen SB, Hellsten Y, Hardie DG, Richter EA (2002) Glycogen-dependent effects of 5-aminoimidazole-4-carboxamide (AICA)-riboside on AMP-activated protein kinase and glycogen synthase activities in rat skeletal muscle. Diabetes 51:284–292
Hegarty BD, Furler SM, Ye J, Cooney GJ, Kraegen EW (2003) The role of intramuscular lipid in insulin resistance. Acta Physiol Scand 178:373–383
Iglesias MA, Ye JM, Frangioudakis G et al (2002) AICAR administration causes an apparent enhancement of muscle and liver insulin action in insulin-resistant high-fat-fed rats. Diabetes 51:2886–2894
Carling D, Fryer LG, Woods A, Daniel T, Jarvie SL, Whitrow H (2003) Bypassing the glucose/fatty acid cycle: AMP-activated protein kinase. Biochem Soc Trans 31:1157–1160
Acknowledgements
This work was supported by a grant to G. Sweeney from the Canadian Diabetes Association in honour of the late N. D. Land. G. Sweeney is also the recipient of a Canadian Diabetes Association scholarship in honor of the late M. A. Bodington, and R. B. Ceddia holds a Canadian Diabetes Association postdoctoral fellowship in honor of the late N. J. Newell. A. Maida was supported by a National Sciences and Engineering Council of Canada (NSERC) summer studentship. We would like to thank A. Klip (The Hospital for Sick Children, Toronto) for the kind gift of the L6GLUT4myc cells, and M. Wheeler and E. Targonsky (University of Toronto) for the AdipoR1 primers.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ceddia, R.B., Somwar, R., Maida, A. et al. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia 48, 132–139 (2005). https://doi.org/10.1007/s00125-004-1609-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-004-1609-y