
Abstract
This review focuses on three preserved, ancient, biological mechanisms (physical activity, insulin sensitivity, and fat storage). Genes in humans and rodents were selected in an environment of high physical activity that favored an optimization of aerobic metabolic pathways to conserve energy for a potential, future food deficiency. Today machines and other technologies have replaced much of the physical activity that selected optimal gene expression for energy metabolism. Distressingly, the negative by-product of a lack of ancient physical activity levels in our modern civilization is an increased risk of chronic disease. We have been employing a rodent wheel-lock model to approximate the reduction in physical activity in humans from the level under which genes were selected to a lower level observed in modern daily functioning. Thus far, two major changes have been identified when rats undertaking daily, natural voluntary running on wheels experience an abrupt cessation of the running (wheel lock model). First, insulin sensitivity in the epitrochlearis muscle of rats falls to sedentary values after 2 days of the cessation of running, confirming the decline to sedentary values in whole-body insulin sensitivity when physically active humans stop high levels of daily exercise. Second, visceral fat increases within 1 week after rats cease daily running, confirming the plasticity of human visceral fat. This review focuses on the supporting data for the aforementioned two outcomes. Our primary goal is to better understand how a physically inactive lifestyle initiates maladaptations that cause chronic disease.
Similar content being viewed by others


Introduction
Exercise is a treatment to prevent most chronic diseases (Booth and Tseng 1995; US Department of Health and Human Resources 1996). More importantly, the lack of regular exercise or physical inactivity is an “actual” cause of chronic diseases (Blair et al. 1993; Mokdad et al. 2004). The CDC has defined inactivity as “not engaging in any regular pattern of physical activity beyond daily functioning” (Centers for Disease Control and Prevention 2007).
A necessary tenet of medicine is to understand underlying biological mechanism of a disease so that effective treatments are developed and preventive methods can be defined. Unfortunately, there is little scientific evidence detailing the biological effects of reduced physical activity on the development of chronic disease. It is therefore, imperative to better understand these mechanisms to improve medical practice and therefore, improve both quality and length of life. This review will focus on three preserved, ancient, biological mechanisms (physical activity, insulin sensitivity, and fat storage).
Selection for aerobic metabolism increased the yield of ATP from 2 to 3 molecules of ATP by anaerobic means from glucose to 36–38 molecules of ATP per molecule of glucose (Brooks et al. 2005). The selective advantage of an increased aerobic capacity for physical activity, according to Bennett and Ruben (1979), are gathering food rather than being the source of food, territorial defense or invasion, and successful courtship and mating. Thus, through the course of evolution, aerobic metabolic pathways must have been selected to optimize fuel utilization during aerobic physical activity (Booth and Lees 2007; Koch and Britton 2007; Raymond and Segre 2006). The aim of this review is to achieve two tasks. The first aim is to discuss an animal model that employs a sudden decrease in physical activity to sedentary levels as a “systemic metabolic knockdown” model. The purpose of the model is to understand which genes might have been selected to maintain and support physical activity throughout evolution. Our approach is also based upon the notion that nature optimized fuel compartmentalization for survival by employing strategies for energy partitioning, conservation, and utilization in physically active humans and animals. Sedentary lifestyles interfere with the inherited metabolic expectancies of fuel turnover caused by daily physical activity. The second aim is to compare the animal model to the limited existing studies using human models of physical inactivity. The purpose is to highlight metabolic alterations that are caused by our new sedentary lifestyle.
Some of the contributors to our ideas on lack of physical activity as a cause of many chronic diseases
In this section, we briefly site the evolution of papers that drive our quest for the mechanisms that underlie the tremendous increase in many chronic diseases caused by a sedentary lifestyle.
In 1970, Åstrand and Rodahl (1970) wrote, “…in close to 100 percent of the biologic existence of our species has been dominated by outdoor activity. Hunting and foraging for food and other necessities in the wilds have been a condition of human life for millions of years. We are adapted to that style of life…we have ended up in an urbanized, highly technologic society. There is obviously no way to revert to our natural way of life…But with insight into our biologic heritage we may yet be able to modify our current life style. Knowledge of the function of the body at rest, as well as during exercise under various conditions is important as a basis for an optimization of our existence.”
In 1988, Eaton et al. (1988) extended Åstrand’s and Rodahl’s tenet,
“From a genetic standpoint, humans living today are Stone Age hunter-gatherers displaced through time to a world that differs from that for which our genetic constitution was selected. Unlike evolutionary maladaptation, our current discordance has little effect on reproductive success; rather it acts as a potent promoter of chronic illnesses: atherosclerosis, essential hypertension, many cancers, diabetes mellitus, and obesity among others. These diseases are the results of interaction between genetically controlled biochemical processes and a myriad of biocultural influences-lifestyle factors-that include nutrition, exercise, and exposure to noxious substances. Although our genes have hardly changed, our culture has been transformed almost beyond recognition during the past 10,000 years, especially since the Industrial Revolution. There is increasing evidence that the resulting mismatch fosters “diseases of civilization” that together cause 75 percent of all deaths in Western nations, but that are rare among persons whose lifeways reflect those of our preagricultural ancestors.”
From these ideas we wrote in 2000 (Booth et al. 2000),
“…we present the concept that the human genome evolved within an environment of high physical activity. Accordingly, we propose that exercise biologists do not study ‘the effect of physical activity’ but in reality study the effect of reintroducing exercise into an unhealthy sedentary population that is genetically programmed to expect physical activity.”
Types of physical inactivity
For purposes of this paper, reductions in physical activity can be subdivided into three categories: (1) elite athlete to physically active lifestyle (2) physically active lifestyle to sedentary, and (3) sedentary to bed rest. In this review we will focus on the change from an active lifestyle to a sedentary lifestyle. We believe this shift best mimics the rapid maladaptations resulting from the collision between a genome selected on a background of physical activity and a sedentary lifestyle produced by a rapidly changing environment.
The rationale for our animal model designed to study physical inactivity
In order to further our understanding of the above concepts in humans, we applied an existing rodent exercise model. Rats naturally will run in wheels, which we reasoned would closely mimic the intermittent physical activity of hunting and foraging for food (mentioned by Åstrand and Rodahl 1970) and Stone Age hunter-gatherers (referred to by Eaton et al.). Cordain et al. (1998) estimate that recently studied hunter-gatherers expended in their daily physical activity the equivalent of 19 km walking (∼24,000 steps) each day. A current human model of our ancestor’s putative level of physical activity might be the US Amish, who have not adopted most of the technological changes occurring during the 20th century. Amish men and women walk 18,000 and 14,000 steps per day, respectively, (Bassett et al. 2004). In Colorado, the US state with the lowest obesity prevalence, the average man and woman walk 6,700 and 6,400 steps per day, respectively, (Wyatt et al. 2005). These data demonstrate that machines and technology have replaced human labor and largely reduced our daily physical activity levels. We contend that voluntary (natural) running closely approximates the daily living environment that generated the human genome. We next reasoned that a sudden termination of natural physical activity by locking the running wheels would allow us to approximate the loss of physical activity from the level of our ancestors to the level of sedentary adults in the US Investigating this loss of activity is extremely important as the Centers for Disease Control and Prevention (Centers for Disease Control and Prevention 2007) report that 25% of US adults are not active at all in their leisure time.
It is important to note that the animals when wheel locked retain ambulatory cage activity and do not become completely immobile like studies using hindlimb unloading in animals and bed rest in humans as we reasoned that the inactivity in those models is more extreme than in modern lifestyle and do not appropriately mimic the loss of activity levels over the last 100–200 years.
In addition while the term “detraining” is commonly associated with athletes to describe the end of high-intensity training, we use the term “reduced natural physical activity” to specify the cessation of natural physical activity which is different than ceasing endurance exercise training. We have previously shown biochemical differences when rats continuously run daily for 2 h (forced treadmill running) or by intermittent, voluntary wheel running for 2 h/day. Mitochondrial markers in a predominantly type II muscle ∼doubled with daily, continuous 2–h treadmill running (Winder et al. 1974), but only increased 26% with 2 h of intermittent voluntary running (Kump and Booth 2005a, b). Further in type I muscle, forced running for two continuous hours ∼doubles mitochondria (Winder et al. 1974), but daily 2-h running produces no change in mitochondrial concentration (unpublished observation). The intermittency of running closely approximates the animals scurrying around fields in search for food, thus serving as a model to study natural physical activity.
In summary the underlying basis for our model mimics that pattern of physical activity for which genes were selected to optimize metabolism to support that activity pattern (Booth et al. 2002a, b; Booth and Lees 2007; Chakravarthy and Booth 2004); and we are employing the rodent wheel lock model to determine what happens to gene expression (protein levels) and their biological function when physical activity is not engaged beyond daily functioning. The primary goal is to better understand how a physically inactivity lifestyle initiates maladaptations that cause chronic disease.
Results of the rodent wheel lock model
We have published four papers using the rodent wheel lock model. Two of the major biological consequences of the shift from high physical activity to a sedentary condition are: a rapid decrease in skeletal muscle insulin sensitivity and a rapid expansion of intra-abdominal fat storage, factors that also play a part in obesity, the metabolic syndrome, and type 2 diabetes.
Reduction in insulin sensitivity
Rapid reductions in insulin sensitivity upon ceasing a physically active lifestyle
The major purpose of our study (Kump and Booth 2005a) was to elucidate some of the mechanisms by which decreasing the natural physical activity causes decreased insulin sensitivity in skeletal muscle. Four-week-old, Fischer-Brown Norway F1-generation male rats had access to running wheels for 3 weeks, running ∼5 km/day in the third week. Wheels were locked for 5 (WL5), 29 (WL29), or 53 h (WL53); a separate group of rats never had wheel access (sedentary, SED). In this paradigm, WL5 rats represent the healthy control while rats who have their wheel locked for longer periods of time (WL29 and WL53) or who never run (SED) represent the “experimental” groups. Relative to WL5, submaximal, insulin-stimulated 2-deoxyglucose uptake into the epitrochlearis muscle was lower in WL53 and SED (Kump and Booth 2005a), demonstrating a rapid reduction in insulin sensitivity of the predominantly type II epitrochlearis muscle in only 2 days. Along with these findings we witnessed reduced insulin binding, insulin receptor beta-subunit (IRbeta) protein level, submaximal insulin-stimulated IRbeta tyrosine phosphorylation, and glucose transporter-4 protein level in both the WL53 and SED rats when compared to the WL5 and WL29 (Kump and Booth 2005a). In addition, Akt/protein kinase B Ser(473) phosphorylation was lower in WL53 and SED than in WL5 demonstrating that both upstream and downstream insulin signaling was reduced following the cessation of daily running (Kump and Booth 2005a).
These findings confirm two human studies that found the loss of whole-body insulin sensitivity at 38 and 60 h after cessation of endurance training. In a study by Burstein et al. (1985) of highly trained athletes; the metabolic clearance rate of glucose during euglycemic clamps was 15.6, 10.1, and 8.5 ml/kg/min at 12, 60, and 168 h, respectively, of no exercise after their last workout. Metabolic clearance rates reached the values of the sedentary group (not athletically active; 7.8 ml/kg/min) at 60 and 168 h of inactivity. Insulin receptor binding to insulin in young erythrocytes also fell 60-h post-exercise (Burstein et al. 1985). In a later study (Oshida et al. 1991), the rate of insulin-mediated glucose uptake (glucose disposal) in a euglycemic clamp was 9.40, 7.78, 6.82, 7.11 ml/kg/min for athletes at 14, 38, 86, and 144 h after their last exercise bout; no differences existed from non-trained subjects (6.80 ml/kg/min) at 38, 86 or 144 h. Our studies extend the human studies by showing rapid declines in insulin sensitivity in rat epitrochlearis muscle upon cessation of habitual exercise (Kump and Booth 2005a).
The rapid decrease in insulin sensitivity by the cessation of a physically active lifestyle initiates metabolic chronic diseases
Our ideas are based upon extensions of earlier papers. In 1972, Lipman et al. (1972) published that the area under the insulin curve during an oral glucose tolerance test significantly increased on the third day of continuous bed rest in humans. Ten years later, we showed that insulin resistance develops in the mouse soleus muscle after 1 day of hindlimb immobilization (Seider et al. 1982). In 1991, Wendorf and Goldfine (1991) hypothesized, “selective insulin resistance in muscle would have the effect of blunting the hypoglycemia that occurs during fasting but would allow energy storage in fat and liver during feeding. Both of these features could allow hunter-gatherers to have survival advantages during periods of food shortage. However, in sedentary individuals allowed free access to food, this genotype would be disadvantageous; these individuals would become obese with concomitant secondary insulin resistance in fat and liver”. In 1999, Grundy (1999) wrote, “Certainly, obesity and physical inactivity are the dominant causes of insulin resistance, although genetic factors undoubtedly affect its severity.” Obesity and the level of habitual physical activity each accounted for ∼25% of the variance in modulation of insulin-mediated glucose disposal in normal volunteers (Bogardus et al. 1985). Given his data, Reaven in 2001 concluded: “…the importance of habitual physical activity, as distinct from exercise training, in modifying the ability of insulin to mediate glucose disposal should no longer be minimized” (Reaven 2001).
In recent years we have emphasized the issues:
-
The presence of physical inactivity during states of continuous feeding results in a downregulation of the activity phenotype leading to insulin resistance, which is an underlying part of metabolic syndrome (Booth et al. 2002a).
-
Reduced physical activity is associated with a rapid development of insulin resistance (Booth et al. 2002b).
-
Physical activity deficiency plays a key role in the rapid development of insulin resistance (Chakravarthy and Booth 2004).
-
Lower mitochondrial densities in skeletal muscle because of physical inactivity may play a role in triggering metabolic dysfunction (Booth and Lees 2007).
As early as 1988, Reaven (1988) proposed that insulin resistance was the primary defect associated with compensatory hyperinsulinemia in a syndrome he called “Syndrome X” that today is known as the metabolic syndrome. He further indicated, “it seems reasonable to suggest that the various facets of Syndrome X are involved to a substantial degree in the cause and course of the major diseases of civilization” (Reaven 1988). In 2006, Willet et al. (2006) identified overweight and inactivity as “the most direct causes” of the insulin resistance syndrome. In 2007, Shulman’s groups reported data supporting the concept that insulin resistance in skeletal muscle likely preludes any other maladaptation(s) like visceral adiposity and alterations in circulating cytokines thought to cause type 2 diabetes. Petersen et al. (2007) concluded that their data support the hypothesis that insulin resistance in skeletal muscle is due to decreased muscle glycogen synthesis, diverting energy away from muscle glycogen synthesis into increased hepatic de novo lipogenesis. They further contend, “…reversing defects in insulin-stimulated glucose transport in skeletal muscle to reverse insulin resistance in this organ might be the best way to prevent the development of the metabolic syndrome at its earliest stages of development.” (Petersen et al. 2007).
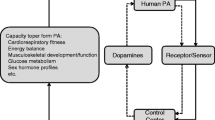
All of the above taken together support our view that a primary consequence of physical inactivity is the development of skeletal muscle insulin resistance, which can contribute to the development of the metabolic syndrome, cardiovascular disease, and type 2 diabetes, as shown in Fig. 1, and described next. Physical inactivity is an actual cause of type 2 diabetes (Mokdad et al. 2004). Primary events from physical inactivity are hypothesized to be decreased utilization of energy-producing substrates by skeletal muscle. As feed-forward events, we hypothesize that skeletal muscle “senses” an underutilization of stored energy in its cells and signals a reduced need for additional uptake of blood glucose, thereby, diminishing insulin sensitivity for glucose uptake. We believe that the aforementioned sequence of events is a remnant of a selected survival mechanism to conserve the limited whole-body stores of carbon atoms for glucose in case a food shortage were to develop (Chakravarthy and Booth 2004; Neel 1962). Such a mechanism would have to be rapid (hours to days) to conserve glucose since body stores of glucose are very limited to <1,000 kcal and can be depleted in less than 1 day. However, in modern humans who live a sedentary lifestyle throughout their lifespan, overt insulin resistance develops contributing to the development of type 2 diabetes. Diabetes has become pandemic, increasing worldwide by fivefold from 1985 to 2000 and the International Diabetes Federation (2007) predicts diabetes to increase another 2.5-fold to 380 million in 2025 (Fig. 2).

A hypothetical sequence to type 2 diabetes is shown (see text for description)

Values taken from the International Diabetes Federation where the year 2025 is their prediction (International Diabetes Federation 2007)
Gain in intra-abdominal fat
Rapid gain in intra-abdominal fat upon ceasing a physically active lifestyle
Rats no longer expend energy in daily running when it ceases. One issue is what happens to the loss of daily energy expenditure? We estimate an excess of 373 kJ (80 and 293 kJ from excess food intake and cessation of daily running) as compared to age-matched cohorts never having access to wheels for running (unpublished estimation) The estimate is the caloric equivalent of 11.5 grams of fat. In our studies of cessation of daily running, we subjectively observed an increase in intra-abdominal fat after 53 h of no running, so we began weighing the fat pads. Cessation of wheel running for 53 h after 21 days of natural running resulted in a 25 and 48% increase in epididymal and omental fat pad weights, respectively (Kump and Booth 2005b).
When humans reduce the number of daily steps from ∼10,000/day to 1,500/day for 2 weeks, intra-abdominal fat mass, as measured by MR-scan, was increased by 7% (Pedersen, unpublished observation). Thus, findings in our experimental animal model of reduced physical activity from naturally high levels to that in a sedentary cage are directionally similar to that in humans decreasing daily stepping to an inactive level.
In another model of inactivity performed in humans where the sedentary group was compared to exercising groups, Slentz et al. (2005) observed a 9% increase in visceral fat over a 6-month period in 40-65 year-old, sedentary (exercise <2 times/week) subjects who were already overweight or mildly obese at the onset of the study (body mass index of 25–35 kg/m2). Two groups exercising at an equivalent of running 12 miles/week at moderate or vigorous intensity prevented the fat gain and a third group exercising at an equivalent of vigorous running 20 miles/week lost 7% abdominal fat (Slentz et al. 2005). Thus, sedentary adults had 16% more visceral fat after 6 months than cohorts who expended calories at a rate equivalent to 20 miles of running/week.
In other randomized controlled clinical trials using imaging techniques to measure changes in visceral fat with exercise (Kay and Fiatarone Singh 2006), the fall in visceral fat ranged anywhere from a 6 to 49% reduction in exercise studies ranging from 8 to 52 weeks in length (Boudou et al. 2003; Irwin et al. 2003; Mourier et al. 1997; Ross et al. 2000, 2004; Weiss and Holloszy 2007). Weight regain is a problem after virtually all dietary and behavioral interventions for obesity (Wadden et al. 2004). The major challenge in the treatment of obesity is maintenance of weight loss (Wing et al. 2006). Regular physical activity has been found in many studies to be associated with long-term weight loss maintenance (Wing and Hill 2001). Most dieters regain about one-third of the weight lost during the next year and between 50 and 97% of individuals return to their baseline in 4–5 years (Kramer et al. 1989; Wadden et al. 1989, 2004; Wing and Hill 2001). Taken together, visceral fat is very plastic in response to changes in the amount of daily physical activity, increasing during times of low activity and decreasing with enhancements in activity.
Cessation of daily running was associated with increased palmitate incorporation into triacylglycerol
The rate of palmitate incorporation in epididymal fat was 4.2-fold greater in SED5 than in WL5, and increased 14-fold between WL5 and WL10, being 79% lower in SED10 than in WL10 (Fig. 3). Palmitate incorporation (index of triacylglycerol synthesis) remained at this elevated level (at least 3.5-fold greater in WL29 and WL53 than SED5). Thus, the rapid increase in epididymal fat mass with the cessation of voluntary wheel running in our model was associated with a prolonged overshoot in palmitate incorporation (Kump and Booth 2005b). The overshoot of palmitate incorporation into triacylglycerol is a training effect of repeated days of running as overshoot did not occur after a single day of running (Kump et al. 2006).

We hypothesize that the provision of wheels in cages allows rats to express their intrinsic drive for natural daily running, exposing the normal physiology of substrate utilization cycling. Removal of daily running inhibits substrate cycling in the ensuing days. Thus, the cessation of daily running stalls the up and down cycle of palmitate incorporation into triacylglycerol in epididymal fat that occurs with daily activity and assumedly leads to an accumulation of adipose tissue. The gray horizontal bar represents their range for palmitate incorporation into triacylaglycerol of age-matched cohorts that never ran; the range of the bar is from 11 am (395 ± 71 pmol per mg homogenate protein per min) to 4 pm (226 ± 35). For more detail, see references (Chakravarthy and Booth 2004; Kump and Booth 2005b)
Mitochondrial glycerol-3-phosphate acyltransferase-1 (mtGPAT1) contributes to the overshoot in palmitate incorporation into triacylglycerol in adipose tissue upon the cessation of daily, natural physical activity
In order to begin to understand the underlying basis for the increased palmitate incorporation into triacylglycerol in intra-abdominal fat upon the cessation of running, we assayed some of the enzymatic steps in triacylglycerol synthesis. Ten hours after 21 days of wheel running, activity of mtGPAT1, a key regulator of triacylglycerol synthesis in epididymal fat, overshot sedentary values by 48% and remained higher than sedentary values at 29 and 53 h of reduced physical activity (Kump et al. 2006). The overshoot in mtGPAT1 activity was accompanied by an increase in mtGPAT protein level with no change in mtGPAT1 mRNA. More than one bout of daily physical activity is necessary to overshoot sedentary values for palmitate incorporation into triacylglycerol in epididymal fat (Kump et al. 2006), which suggests that repeated days of physical activity (chronic effect) are responsible for this overshoot effect. We conclude that an increase in mtGPAT protein in epididymal fat associates with the overshoot in palmitate incorporation in epididymal fat; this adaptation would enhance restoration of fat stores after cessation of daily physical activity, enhancing survival in case a food deficiency were to occur.
Adipocyte hyperplasia is accentuated after the cessation of daily wheel running
In order to determine if increased intra-abdominal fat is solely due to increased food consumption, and not a cessation of exercise, we tested the hypothesis that reduced physical activity, independent of excessive caloric intake, would be unable to induce an increase in fat pad mass. The experimental design was modified from our three studies (Kump and Booth 2005a, b; Kump et al. 2006), above. 21-day-old rats were given access to voluntary running wheels for 42–43 days, thus starting the rats running at a younger age and having them run twice as long before locking the wheels. Rats were running approximately 9 km/day in the last week, after which wheels were locked for 5, 53, or 173 h (WL5, WL53, WL173) before sacrifice. During the 53 and 173 h of inactivity, one group of animals was pair fed (PF) to match sedentary controls, whereas the other continued to eat ad libitum (AL). Epididymal and retroperitoneal fat masses were significantly increased in the WL173-PF versus the WL5 group, whereas epididymal, perirenal, and retroperitoneal fat masses were all significantly increased in the WL173-AL group compared with the WL5 group (Laye et al. 2007). The demonstrated increase in intra-abdominal fat stores after the cessation of running in the absence of hyperphagia demonstrates that physical inactivity induced fat storage is independent of food intake. Additionally, hyperplasia of adipocytes in epididymal fat mass occurred in WL173-AL but no change in mean adipocyte size was noted.
Interim summary of our animal model of reduced physical activity
Two major changes have been identified when 21- or 28-day-old rats undertake 3 or 6 weeks of daily, natural voluntary running on wheels, followed by an abrupt cessation of the running. First, insulin sensitivity in the epitrochlearis muscle of rats falls to sedentary values with 2 days of the cessation of running (Kump and Booth 2005a), confirming the decline to sedentary values in whole-body insulin sensitivity when physically active humans stop daily exercise (Burstein et al. 1985; Oshida et al. 1991). Second, visceral fat increases within 1 week when rats cease daily running (Laye et al. 2007), confirming the plasticity of human visceral fat. A fundamental question in biology then becomes, why were rats and humans built this way?
Speculated bases for the inactivity-induced changes
Why is the body so plastic to decreases in physical activity?
One answer to the question was popularized by Neel (1962) who in 1962 introduced the concept of “thrifty” genotype. Neel (1962) proposed that certain genotypes were selected into the human genome because of their selective advantage over the less “thrifty” ones. Neel (1962) defined a “thrifty” genotype as “being exceptionally efficient in the intake and/or utilization of food”. The rationale is that during periods of food deprivation, animals and humans with the “thrifty” genotype would have a survival advantage because they could rely on larger, previously stored energy to maintain homeostasis, whereas those without “thrifty” genotypes would be at a disadvantage by having smaller fuel stores at the start of the food deprivation, and therefore, would be less likely to survive (Neel 1962). Chakravarthy and Booth (2004) extended Neel’s thrifty genotype by postulating that survival during the feast-food deprivation cycle of the hunter-gatherer also selected genes to support a “physical activity cycle” in which cycling of metabolic processes was triggered by the reduction of skeletal muscle glycogen and triglyceride stores. Indeed, our recent work provides an example of such speculation.
Cessation of daily, natural physical activity is associated with a disappearance of the natural cycling of palmitate incorporation (an index of triacylglycerol synthesis) into triacylglycerol of epididymal fat (Fig. 3). To speculate on the potential biological significance of triacylglycerol cycling, we will apply concepts of evolutionary or Darwinian medicine (Williams and Nesse 1991; Eaton et al. 2002) to understand from an evolutionary perspective why the human body is not better designed for physical inactivity. We suggest that increased fat storage upon initiating physical inactivity is an inherited survival mechanism to store energy as fat in anticipation of a future food deficiency; it accomplishes this feat by imposing decreased insulin sensitivity to repartition glucose from inactive skeletal muscle to adipocytes, where energy storage occurs. The phenotype becomes maladaptive when physical inactivity continues unabated for months/years.
To sum up our beliefs as to why natural selection resulted in energy partitioning from inactive skeletal muscle to fat is energy conservation so that fuels not needed for work are stored for survival in the next food deficiency.
Rationale for rapidity of changes to reductions in enhanced physical activity
Genes that regulate PDK4 mRNA/protein degradation were selected into the human genome
As the onset of food deficiency is rapid, adaptations for energy storage and utilization to enhance the probability of survival had to be rapid, leading to the need to have a rapid turnover proteins involved in energy metabolism. Important regulatory processes must, therefore be very plastic. Natural selection is based upon the concept that those organisms that can adapt to a new environment best are more likely to pass their gene pools on to the next generation as compared to those with slower adaptations to the new environment. Therefore, one criterion for selection of genes to study function when reduced inactivity ensues would be to consider whether their half life was short. Goldberg and Dice (1974) has commented that of the 40 liver proteins that have the shortest half lives [determined by (Berlin and Schimke 1965)] were either rate limiting or first steps in pathways. Whereas, the twelve proteins that had the longest half lives did not function as rate limiting. Thus, principles of protein turnover will be briefly described.
The importance of short half lives (t½) is that the time course between steady state levels of protein is determined solely by the first-order rate constant for degradation (k d), where t½ = ln 2/K d (Schimke 1970). Thus, proteins with shorter half lives adapt to new steady state levels faster than proteins with longer half lives challenged by an environmental perturbation (Schimke 1970). Natural selection is based upon the concept that those organisms that can adapt to a new environment best are more likely pass their gene pools on to the next generation as compared to those with slower adaptations to the new environment. Therefore, one criterion for selection of genes to study when reduced inactivity ensues would be to consider whether the half life of the protein they encoded for was short.
One such example of a key regulatory protein is pyruvate dehydrogenase kinase 4 (PDK4). PDK4, when activated, phosphorylates and inactivates the pyruvate dehydrogenase complex (PDC). As PDC is switched between glucose-derived pyruvate and long chain fatty acid entry into the TCA cycle, increased PDK4 activity would inhibit glycolysis, conserving glucose carbon chains from oxidation. PDK4 has a very short half life of 1.3 h in Morris heptoma 7800 C1 cells (Huang et al. 2002). Two hours following an endurance exercise bout, PDK4 mRNA increases ∼fourfold (Pilegaard et al. 2005). Further at 8 h post-exercise, PDK4 mRNA had returned to pre-exercise values in muscles having high carbohydrate levels before exercise as opposed to muscles with low carbohydrate at the start of exercise (Pilegaard et al. 2005), demonstrating the importance of local energy status in regulating its expression. Regulation of PDK4 mRNA has been shown at the transcriptional level; no data is yet available that shows regulation by its mRNA stability (Sugden and Holness 2006). However, since the rise in PDK4 mRNA and protein is very transient after a single bout of exercise, specific mechanisms, yet unknown, must have been selected to regulate the rapid increase in its transcription and its rapid decrease in mRNA and/or protein. In conclusion, the putative function of high PDK4 protein (inferred from its high mRNA) during exercise would be to conserve the limited stores of whole-body glucose carbons for the central nervous system and red blood cells, which do not oxidize fatty acids. Further, fast turnover of molecules allows rapid adaptive changes to meet new metabolic challenges.
Concluding remarks
Current human biology is a prisoner of the metabolic pathways designed by our evolution. Without knowledge of selected genes and their functions for which they evolved, a precise definition of normal function is impossible. Less than adequate knowledge leads to incorrect interpretations and less than optimal preventative measures and therapies to improve health. The selection of genes for survival over hundreds of thousands of years includes those metabolic processes that were optimized for energy partitioning, conservation, and utilization. Genes were selected to allow aerobic metabolism and to orchestrate the complex integration needed for physical activity for survival in food acquisition, defense, and reproduction. Imposition of minimal daily physical activity on a system primed for aerobic activity initially results in fuel redistribution to minimize glucose uptake into skeletal muscle and to maximize fat storage and eventually progresses to obesity and type 2 diabetes.
References
Åstrand PO, Rodahl K (1970). Textbook of work physiology. McGraw-Hill, New York
Bassett DR, Schneider PL, Huntington GE (2004) Physical activity in an Old Order Amish community. Med Sci Sports Exerc 36:79–85
Bennett AF, Ruben JA (1979) Endothermy and activity in vertebrates. Science 206:649–654
Berlin CM, Schimke RT (1965) Influence of turnover rates on the responses of enzymes to cortisone. Mol Pharmacol 1:149–156
Blair SN, Powell KE, Bazzarre TL, Early JL, Epstein LH, Green LW, Harris SS, Haskell WL, King AC, Koplan J (1993) Physical inactivity. Workshop V. AHA prevention conference III. Behavior change and compliance: keys to improving cardiovascular health. Circulation 88:1402–1405
Bogardus C, Lillioja S, Mott DM, Hollenbeck C, Reaven G (1985) Relationship between degree of obesity and in vivo insulin action in man. Am J Physiol 248:E286–E291
Booth FW, Lees SJ (2007) Fundamental questions about genes, inactivity, and chronic diseases. Physiol Genomics 28:146–157
Booth FW, Tseng BS (1995) America needs to exercise for health. Med Sci Sports Exerc 27:462–465
Booth FW, Gordon SE, Carlson CJ, Hamilton MT (2000) Waging war on modern chronic diseases: primary prevention through exercise biology. J Appl Physiol 88:774–787
Booth FW Chakravarthy MV, Gordon SE, Spangenburg EE (2002a) Waging war on physical inactivity: using modern molecular ammunition against an ancient enemy. J Appl Physiol 93:3–30
Booth FW, Chakravarthy MV, Spangenburg EE (2002b) Exercise and gene expression: physiological regulation of the human genome through physical activity. J Physiol 543:399–411
Boudou P, Sobngwi E, Mauvais-Jarvis F, Vexiau P, Gautier JF (2003) Absence of exercise-induced variations in adiponectin levels despite decreased abdominal adiposity and improved insulin sensitivity in type 2 diabetic men. Eur J Endocrinol 149:421–424
Brooks GA, Fahey TD, Baldwin KM (2005) Exercise physiology. McGraw-HIll, New York
Burstein R, Polychronakos C, Toews CJ, MacDougall JD, Guyda HJ, Posner BI (1985) Acute reversal of the enhanced insulin action in trained athletes. Association with insulin receptor changes. Diabetes 34:756–760
Centers for Disease Control and Prevention (2007) Physical activity terms
Chakravarthy MV, Booth FW (2004) Eating, exercise, and “thrifty” genotypes: connecting the dots toward an evolutionary understanding of modern chronic diseases. J Appl Physiol 96:3–10
Cordain L, Gotshall RW, Eaton SB, Eaton SB III (1998) Physical activity, energy expenditure and fitness: an evolutionary perspective. Int J Sports Med 19:328–335
Eaton SB, Konner M, Shostak M (1988) Stone agers in the fast lane: chronic degenerative diseases in evolutionary perspective. Am J Med 84:739–749
Eaton SB, Strassman BI, Nesse RM, NEEL JV, Ewald PW, Williams GC, Weder AB, Eaton SB III, Lindeberg S, Konner MJ, Mysterud I, Cordain L (2002) Evolutionary health promotion. Prev Med 34:109–118
Goldberg AL, Dice JF (1974) Intracellular protein degradation in mammalian and bacterial cells. Annu Rev Biochem 43:835–869
Grundy SM (1999) Primary prevention of coronary heart disease: integrating risk assessment with intervention. Circulation 100:988–998
Huang B, Wu P, Bowker-Kinley MM, Harris RA (2002) Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes 51:276–283
International Diabetes Federation (2007) Diabetes prevalence http://www.idf.org/home/index.cfm?node=264
Irwin ML, Yasui Y, Ulrich CM, Bowen D, Rudolph RE, Schwartz RS, Yukawa M, Aiello E, Potter JD, McTiernan A (2003) Effect of exercise on total and intra-abdominal body fat in postmenopausal women: a randomized controlled trial. JAMA 289:323–330
Kay SJ, Fiatarone Singh MA (2006) The influence of physical activity on abdominal fat: a systematic review of the literature. Obes Rev 7:183–200
Koch LG, Britton SL (2007) Evolution, atmospheric oxygen, and complex disease. Physiol Genomics 30:205–208
Kramer FM, Jeffery RW, Forster JL, Snell MK (1989) Long-term follow-up of behavioral treatment for obesity: patterns of weight regain among men and women. Int J Obes 13:123–136
Kump DS, Booth FW (2005a) Alterations in insulin receptor signalling in the rat epitrochlearis muscle upon cessation of voluntary exercise. J Physiol 562:829–838
Kump DS, Booth FW (2005b) Sustained rise in triacylglycerol synthesis and increased epididymal fat mass when rats cease voluntary wheel running. J Physiol 565:911–925
Kump DS, Laye MJ, Booth FW (2006) Increased mitochondrial glycerol-3-phosphate acyltransferase protein and enzyme activity in rat epididymal fat upon cessation of wheel running. Am J Physiol Endocrinol Metab 290:E480–E489
Laye MJ, Thyfault JP, Stump CS, Booth FW (2007) Inactivity induces increases in abdominal fat. J Appl Physiol 102:1341–1347
Lipman RL, Raskin P, Love T, Triebwasser J, Lecocq FR, Schnure JJ (1972) Glucose intolerance during decreased physical activity in man. Diabetes 21:101–107
Mokdad AH, Marks JS, Stroup DF, Gerberding JL (2004) Actual causes of death in the United States, 2000. JAMA 291:1238–1245
Mourier A, Gautier JF, De KE, Bigard AX, Villette JM, Garnier JP, Duvallet A, Guezennec CY, Cathelineau G (1997) Mobilization of visceral adipose tissue related to the improvement in insulin sensitivity in response to physical training in NIDDM. Effects of branched-chain amino acid supplements. Diabetes Care 20:385–391
Neel JV (1962) Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14:353–362
Oshida Y, Yamanouchi K, Hayamizu S, Nagasawa J, Ohsawa I, Sato Y (1991) Effects of training and training cessation on insulin action. Int J Sports Med 12:484–486
Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, Yonemitsu S, Cline GW, Befroy D, Zemany L, Kahn BB, Papademetris X, Rothman DL, Shulman GI (2007) Inaugural Article: The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA 104:12587–12594
Pilegaard H, Osada T, Andersen LT, Helge JW, Saltin B, Neufer PD (2005) Substrate availability and transcriptional regulation of metabolic genes in human skeletal muscle during recovery from exercise. Metabolism 54:1048–1055
Raymond J, Segre D (2006) The effect of oxygen on biochemical networks and the evolution of complex life. Science 311:1764–1767
Reaven GM (1988) Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37:1595–1607
Reaven GM (2001) Insulin resistance, compensatory hyperinsulinemia, and coronary heart disease: syndrome X revisited. In: Jefferson LS, Cherrington AD (eds) Handbook of physiology. Section 7: the endocrine system. Vol II: the endocrine pancreas and regulation of metabolism. Oxford University Press, New York, pp 1169–1197
Ross R, Dagnone D, Jones PJ, Smith H, Paddags A, Hudson R, Janssen I (2000) Reduction in obesity and related comorbid conditions after diet-induced weight loss or exercise-induced weight loss in men. A randomized, controlled trial. Ann Intern Med 133:92–103
Ross R, Janssen I, Dawson J, Kungl AM, Kuk JL, Wong SL, Nguyen-Duy TB, Lee S, Kilpatrick K, Hudson R (2004) Exercise-induced reduction in obesity and insulin resistance in women: a randomized controlled trial. Obes Res 12:789–798
Schimke RT (1970) Regulation of protein degradation in mammalian tissues. In: Munto HN (eds) Mammalian protein metabolism. Academic, New York, pp 177–228
Seider MJ, Nicholson WF, Booth FW (1982) Insulin resistance for glucose metabolism in disused soleus muscle of mice. Am J Physiol 242:E12–E18
Slentz CA, Aiken LB, Houmard JA, Bales CW, Johnson JL, Tanner CJ, Duscha BD, Kraus WE (2005) Inactivity, exercise, and visceral fat. STRRIDE: a randomized, controlled study of exercise intensity and amount. J Appl Physiol 99:1613–1618
Sugden MC, Holness MJ (2006) Mechanisms underlying regulation of the expression and activities of the mammalian pyruvate dehydrogenase kinases. Arch Physiol Biochem 112:139–149
US Department of Health and Human Resources (1996) Physical Activity and Health: A Report of the Surgeon General. US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Atlanta, GA
Wadden TA, Sternberg JA, Letizia KA, Stunkard AJ, Foster GD (1989) Treatment of obesity by very low calorie diet, behavior therapy, and their combination: a five-year perspective. Int J Obes 13(Suppl 2):39–46
Wadden TA, Butryn ML, Byrne KJ (2004) Efficacy of lifestyle modification for long-term weight control. Obes Res 12(Suppl):151S–162S
Weiss EP, Holloszy JO (2007) Improvements in body composition, glucose tolerance, and insulin action induced by increasing energy expenditure or decreasing energy intake. J Nutr 137:1087–1090
Wendorf M, Goldfine ID (1991) Archaeology of NIDDM. Excavation of the “thrifty” genotype. Diabetes 40:161–165
Willet WC, Koplan JP, Nugent R, Dusenbury C, Puska P, Gaziano TA (2006) Prevention of chronic disease by means of diet and lifestyle changes. In: Jamison DT, Breman JG, Measham, Measham AR, Alleyne G, Evans DB, Jha P, Mills A, Musgrove P (eds) Disease control priorities in developing countries. World Bank, pp 833–850
Williams GC, Nesse RM (1991) The dawn of Darwinian medicine. Q Rev Biol 66:1–22
Winder WW, Baldwin KM, Holloszy JO (1974) Enzymes involved in ketone utilization in different types of muscle: adaptation to exercise. Eur J Biochem 47:461–467
Wing RR, Hill JO (2001) Successful weight loss maintenance. Annu Rev Nutr 21:323–341
Wing RR, Tate DF, Gorin AA, Raynor HA, Fava JL (2006) A self-regulation program for maintenance of weight loss. N Engl J Med 355:1563–1571
Wyatt HR, Peters JC, Reed GW, Barry M, Hill JO (2005) A Colorado statewide survey of walking and its relation to excessive weight. Med Sci Sports Exerc 37:724–730
Acknowledgments
The authors thank the University of Missouri Research Board (FB), the College of Veterinary Medicine Research Fund (FB), and the Life Sciences Predoctoral Fellowship Program (ML) for support of the reported research. The review was written while supported by Department of Veterans Affairs Career Development Grant—CDA-2 (JPT) and Department of Internal Medicine at University of Missouri (RSR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Booth, F.W., Laye, M.J., Lees, S.J. et al. Reduced physical activity and risk of chronic disease: the biology behind the consequences. Eur J Appl Physiol 102, 381–390 (2008). https://doi.org/10.1007/s00421-007-0606-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00421-007-0606-5