
Abstract
The outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) in December 2019 form Wuhan, China leads to coronavirus disease 2019 (COVID-19) pandemic. While the common cold symptoms are observed in mild cases, COVID-19 is accompanied by multiorgan failure in severe patients. The involvement of different organs in severe patients results in lengthening the hospitalization duration and increasing the mortality rate. In this review, we aimed to investigate the involvement of different organs in COVID-19 patients, particularly in severe cases. Also, we tried to define the potential underlying mechanisms of SARS-CoV2 induced multiorgan failure. The multi-organ dysfunction is characterized by acute lung failure, acute liver failure, acute kidney injury, cardiovascular disease, and as well as a wide spectrum of hematological abnormalities and neurological disorders. The most important mechanisms are related to the direct and indirect pathogenic features of SARS-CoV2. Although the presence of angiotensin-converting enzyme 2, a receptor of SARS-CoV2 in the lung, heart, kidney, testis, liver, lymphocytes, and nervous system was confirmed, there are controversial findings to about the observation of SARS-CoV2 RNA in these organs. Moreover, the organ failure may be induced by the cytokine storm, a result of increased levels of inflammatory mediators, endothelial dysfunction, coagulation abnormalities, and infiltration of inflammatory cells into the organs. Therefore, further investigations are needed to detect the exact mechanisms of pathogenesis. Since the involvement of several organs in COVID-19 patients is important for clinicians, increasing their knowledge may help to improve the outcomes and decrease the rate of mortality and morbidity.
Similar content being viewed by others

Introduction
Human coronaviruses (HCoVs) are associated with multiple respiratory diseases (Lim et al. 2016). Up to now, the identified HCoVs, including HCoV229E, HCoV-OC43, HCoV-HKU1, and HCoV-NL63 are related to the mild disease. Also, the 2002-4 epidemic due to severe acute respiratory syndrome coronavirus (SARS-CoV) and the 2012 epidemic due to Middle East respiratory syndrome coronavirus (MERS-CoV) were considered as members of the HCoVs with mild to severe consequences and death. In December 2019, the coronavirus disease-2019 (COVID-19) pandemic as a result of a novel virus outbreak, SARS-CoV2 from Wuhan, China (Liu et al. 2020a). This virus is categorized as a beta-CoV subgroup which belongs to the Coronaviridae family (Sahu et al. 2020a). The transmission of through respiratory droplets, aerosol, and mucosal membrane contact with fomites were confirmed (Rothan and Byrareddy 2020; Van Doremalen et al. 2020). Also, the SARS-CoV2 RNA was identified in the stools of COVID-19 patients (Zhang et al. 2020d).
In a large study, severe illness was seen in 15.7% of COVID-19 patients following hospitalization, 5% of these patients were admitted to ICU, 2.3% required intubation, and 1.4% died (Baud et al. 2020). A broad range of manifestations from asymptomatic to severe has been defined for COVID-19 (Lei et al. 2020). In general, patients with exposure to respiratory CoVs show the common cold manifestations, bronchiolitis, and pneumonia (Cabeça et al. 2013). The clinical manifestations in mild COVID-19 patients are fever, dry cough, and myalgia (fatigue) (Lei et al. 2020). While the high rate of patients are asymptomatic or show the favorable prognosis, the worse outcomes were seen in old patients and also cases with underlying condition e.g., cardiovascular disease (CVD), renal disease, diabetes, high blood pressure, malignancy, obesity, and chronic respiratory diseases (Guzik et al. 2020). The high mortality in COVID-19 was seen in patients with these underlying conditions (Alberici et al. 2020). One week after onset of disease, COVID-19 can progress as a severe condition with hypoxemia and dyspnea, and rapidly develop to acute respiratory distress syndrome (ARDS) (Cheng et al. 2020a). COVID-19 also is associated with the involvement of different organs and systems, such as lung, liver, kidney, heart, and gastrointestinal, hematological, and nervous system with a high rate of mortality and induction of multi-organ failure in infected patients (Gupta et al. 2020). Up to now, no specific treatment for severe illness or vaccines against SARS-CoV2 particularly has not been introduced (Fauci et al. 2020).
In this review, we investigated the multi-organ failure and related symptoms induced by SARS-CoV2 in severe patients. Also, we described the potential molecular mechanisms of this virus in the induction of pathologic features.
COVID-19: General features
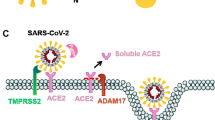
SARS-CoV2, an enveloped, non-segmented, and positive-sense RNA virus, belongs to the β-CoVs (Li et al. 2020a). The complete genome of SARS-CoV2 isolated from a patient was 29.9 kb (Liu et al. 2020d) and many similarities were identified in the genome of SARS-CoV2 with SARS-CoV (79.5%) as a bat-origin CoV (Andersen et al. 2020). A part of the genome is translated into four structural proteins, including spike (S) glycoprotein, matrix (M), and nucleocapsid (N), and small envelope (E) proteins. These proteins can stimulate the host immune system (Naqvi et al. 2020). S-protein of virus binds to the ACE2 receptor to enter into the host cells (Diaz 2020). RBD which mainly be distributed in the respiratory system (Diaz 2020). The mutation of SARS-CoV2 has enhanced the affinity of this virus to ACE2 in human and decreased in rodents (Yang et al. 2020c). Both SARS-CoV and SARS-CoV2 use ACE2 as a receptor to invade the host cells (Wu et al. 2020d). ACE2 is a type I transmembrane protein with 805 amino acids that generate angiotensin (Ang)-II, an important mediator of renin-Ang system (Verdecchia et al. 2020). When the virus enters into the cell, it releases the RNA into the cytoplasm, translated into two virus polyproteins and structural proteins. The genome of the virus then begins to replicate and forms a nucleocapsid in combination with genomic RNA and nucleocapsid protein (Perlman and Netland 2009). Then, viral particles use the cellular protein synthesis organelles, such as Endoplasmic reticulum and Golgi complex to be germinated. Finally, vesicles including the viral particles bind to the plasma membrane to be released (De Wit et al. 2016). The infection with SARS-CoV2 commonly leads to fever, fatigue, dry cough, anorexia, and dyspnea (Lei et al. 2020; Wang et al. 2020a). In severe cases, the presence of ACE2 receptor on the cells of different organs may lead to organ dysfunction (Zhang et al. 2020e).
COVID-19 and lung diseases
Features of lung diseases in COVID-19
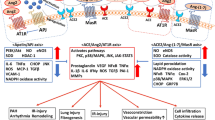
Pneumonia is the most serious symptom of COVID-19, featured by cough, fever, breath shortness, chest pain, dyspnea, fatigue, and bilateral diffuse interstitial pattern on x-ray chest image. This indicates that there is no specific clinical feature to distinguish COVID-19 from other viral respiratory diseases (Mukherjee et al. 2020; Yi et al. 2020). These manifestations are commonly observed in several respiratory and other diseases (such as influenza, common colds, asthma, and allergies). Therefore, the clinician can get confused with the other common diseases presenting with the same features (Sahu et al. 2020b). One week after onset of illness, this condition could develop as ARDS in 17% of patients and 65% of these patients worsened and died due to multi-organ dysfunction (Chen et al. 2020a). The incidence of ARDS was reported 15.6 to31%, higher than that of other organ impairment (Li and Ma 2020). ARDS has been demonstrated to be strongly linked with the older age group (more frequent in > 65 years old), hypertension, and diabetes mellitus (Wu et al. 2020b). The SARS-CoV2 infection demonstrates similar radiological properties with other respiratory diseases (Robles et al. 2011). The computed tomopraphy (CT) findings exposed ground-glass opacities, consolidative opacities, as well as crazy paving patterns in both lungs (Chung et al. 2020) A staging system for using CT images has been reported (Jin et al. 2020). Notably, the progressive pleural effusions were reported in late-stages (Shi et al. 2020a) (Fig. 1).

The features of multiorgan failure were summarized due to the laboratory and imaging findings. hs-cTnI sensitive cardiac troponin I, CK creatine cinase, α-HBDH α-hydroxybutyrate dehydrogenase, LV left ventricular, ALT alanine aminotransferase, AST aspartate aminotransferase, TB total bilirubin, GGT gamma-glutamyl transferase, ALP alkaline phosphatase, Cr creatinin, eGFR estimated glomerular filtration rate, BUN blood urea nitrogen, DIC disseminated intravascular coagulation, VTE venous thromboembolism, LDH lactate dehydrogenase, NLR neutrophil/lymphocyte ratio, PLR platelet/lymphocyte ratio, PT prothrombin time, aPTT activated partial thromboplastin time
Mechanisms of SARS-CoV2 -induced lung diseases
SARS-COV2 infection may induce the injury in the lung through direct and indirect pathways. While ACE2 exerts protective features against the ALF progression (Imai et al. 2005), it is a functional receptor for SARS-CoV and SARS-CoV2 entry (Hamming et al. 2004). Attachment of the virus to ACE2 and invasion of host cells can directly infect them and increase the inflammatory responses and consequently lead to the cell death and lung complications (Verdecchia et al. 2020). ACE2 is a cell surface protein, located on the upper airway (goblet and ciliated epithelial cells), lower respiratory tract epithelium (Type II alveolar), and pulmonary vasculature, such as arterial smooth muscle, as well as endothelial cells of veins and arteries (Gheblawi et al. 2020; Imai et al. 2005; Li 2016). The high distribution of ACE2 was recognized on type II alveolar cells of the human lung (Hamming et al. 2004). Inhibition of ACE2 was shown to have a critical role in the control of SARS-CoV infection induced-pathological alteration of the lung which leads to ALF and severe pneumonia (Pyrc et al. 2007). In a murine model, S-protein of SARS-CoV binds to ACE2 and results in pulmonary edema and decreased lung function caused by increased pulmonary vascular permeability induced by Ang-II activation (Imai et al. 2005).
Notably, the infection with SARS-CoV2 results in interstitial inflammation and alveolar injury. The pathologic features of ARDS are also related to the activation of Ang-II pathways which interferes with adaptive immunity through the stimulation of macrophages and other immune cells (Bernstein et al. 2018) and enhanced concentrations of interleukin (IL)-6, tumor necrosis factor-alpha (TNF-α), and other inflammatory cytokines (Lee et al. 2002; Yamamoto et al. 2011). The activated dendritic cells and macrophages enhance the phagocytosis of the apoptotic virus-infected epithelial cells (Nainu et al. 2017). The enhanced levels of pro-inflammatory mediators e.g., interferon-gamma (IFNγ), ILs (1β, 6, and 12) and chemokines e.g., CXC L10, and CCL2, well-known as “cytokine storm” and subsequent “hyperinflammation syndrome”, were confirmed in pulmonary inflammation, induced by SAR-CoV, MERS-CoV, SARS-CoV2 infections (Channappanavar and Perlman 2017; Lei et al. 2020). Higher levels of CXCL10, CCL2, and TNFα were recorded in COVID-19 patients admitted to ICU severe compared to the other cases. Also, increased cytokines, produced by CD4+ T-helper 2 e.g., IL-4 and IL-10 with inflammation suppressive features, were observed in these patients (Zhang et al. 2020c). The cytokine storm, followed by the attacking immune system via SARS-CoV2, results in lung failure, multiple-organ dysfunction, and death in severe cases (Xu et al. 2020b). Therefore, the pathologic characteristics of ARDS with alveolo-capillary membrane injuries as a result of enhanced permeability of lung and permeation of edema fluid into the airspaces and subsequent respiratory insufficiency were related to the cytokine storm (Bhatia et al. 2012). The activation of the complement system, the mediator of acute and chronic inflammation was found in the post-mortem lung tissue of COVID-19 patient. The components, including C4, C3, C5b-9, and mannose-binding lectin, were positive in alveolar epithelial cells, some pneumocytes, immune cells, and exudates in alveolar spaces (Gao et al. 2020).
The recent study demonstrated that ARDS is related to the obstructive thromboinflammatory syndrome (MicroCLOTS) in the microvascular lung vessels of COVID-19 patients (Ciceri et al. 2020). In a case-series study, the haemorrhage and/or thromboemboli in the major pulmonary vessels were reported in patients with COVID-19 (Barton et al. 2020; Xu et al. 2020b). Also, the pulmonary CT angiography has proven that COVID-19 pneumonia is related to acute pulmonary embolism (Grillet et al. 2020). The autopsy evaluations of lung tissue from a COVID-19 patient demonstrated the histopathological features, including acute interstitial pneumonia, diffuse alveolar damages with perivascular infiltration of macrophages and T-cell, disrupted cell membrane (direct SARS-CoV2 infection), intussusceptive angiogenesis, the formation of hyaline membranes, and oedema in the alveolar wall. The microvasculature involvement, such as endothelial damage, hyaline thrombosis in pulmonary vessels (intra and extra), vessel wall oedema, intravascular neutrophil traps, and infiltration of inflammatory cells (Ackermann et al. 2020; Barton et al. 2020). The formation of microthrombi within the vascular structure of pulmonary tissue leads to pulmonary hypertension, pulmonary haemorrhage, pulmonary infarction, and secondary ventricular stress (Wölfel et al. 2020). Taken together, the direct infection of the lung by virus, cytokine storm, and microthrombi formation should be considered as underlying mechanisms of pulmonary failure induced by SARS-CoV2 infection.
COVID-19 and heart diseases
Features of heart diseases in COVID-19
Viral infections cause myocarditis in developed countries which are considered as a reason for acute heart failure, chronic dilated cardiomyopathy, and sudden death (Sagar et al. 2012). Some viruses e.g., enterovirus adenovirus and herpesvirus are commonly seen in the etiologies for CVDs (Pollack et al. 2015). In several studies have been shown that coronavirus is a certain pathogen for the heart and induces myocarditis in the infected patients (Alhogbani 2016). In the recent pandemic, the cardiovascular complications, such as cardiac injury (12%) (Yang et al. 2020b) and arrhythmias (44%) were reported in SARS-CoV2 infected cases (Wang et al. 2020c). In a case report study, it has been demonstrated that MERS-CoV infection creates acute myocarditis, and results in acute myocardial injury and myocardial edema in lateral and apical walls of the left ventricle (Shi et al. 2020b). Similarly, infected patients with the SARS-CoV demonstrated CVD, from systolic and diastolic abnormalities to arrhythmias (Oudit et al. 2009b). Likewise, a single-center study showed that 40% of patients with COVID-19 (mean age; 55.5 years) had underlying conditions, such as CVD (Chen et al. 2020b). Additionally, in a comprehensive analysis of six clinical studies on a total number of 783 patients with COVID-19, mortality rate was almost 2% mainly due to ARDS, acute kidney injury (AKI), and myocardial injury. The major complications leading to death probably are related to pre-existing CVD (Burchfield 2020; Jiang et al. 2020).
Myocardial injury was recognized by elevated cardiac biomarkers levels in the first cases in China (Clerkin et al. 2020). High-sensitive cardiac troponin I (hs-cTnI) is a biomarker for the identification of myocardial ischemia and myocardial necrosis, suggesting that an aberrantly elevated hs-cTnI usually is an indicator of myocardial infarction (Hładij et al. 2017; Neumann et al. 2017). In the first study on SARS CoV2 infected patients (n = 41) in Wuhan, five cases were diagnosed with myocardial injury mainly through increased levels of hs-cTnI (> 28 pg/mL) (Huang et al. 2020). In a report, 12% of COVID-19 patients without a history of CVDs demonstrated increased levels of troponin levels or cardiac arrest during hospitalization period (Zheng et al. 2020). The heart injury has been demonstrated by creatine kinase myocardial band (CK-MB) (Kemp et al. 2004; Ternant et al. 2017). The peak levels of CK-MB occurs after 12 h and return to the initial amount within 48–72 h (Ndrepepa and Kastrati 2018). In a study on admitted patients in Wuhan Hospital (n = 188), the male and smoker patients were more susceptible to increased MB-CK levels (Wu et al. 2020c). The levels of myocardial injury biomarkers (CK-MB and hs-cTnI) in patients required to the ICU admission significantly increased compared with patients in the general ward, confirming that severe symptoms indicate on serious complications, including acute myocardial injury (Wang et al. 2020b). The elevated levels of two kinds of myocardial (iso)enzymes, including lactate dehydrogenase (LDH) and α-Hydroxybutyrate dehydrogenase (α-HBDH) indicate on myocarditis or myocardial infarction (Vasudevan et al. 1978). During the hospitalization, the group with severer hemocytopenia had a higher level of LDH compared with the less severe group (Lu et al. 2020). In a similar study, it was revealed that older patients and patients with hypertension were more vulnerable to enhanced levels of LDH and α-HBDH and this condition not only occurs in heart injury, but also was observed in the lung or other organ damages. However, a high level of LDL during the admission was related to reduced survival chance (Wu et al. 2020c). A study on the epidemiological and demographic features, clinical manifestations, laboratory biomarkers, echocardiography, electrocardiography, and chest imaging findings of 102 patients showed that the elevated levels of C-reactive protein (CRP), old age, underlying conditions, and the severity of pneumonia are the most important risk factors for cardiac involvement in COVID-19 patients (Xu et al. 2020c) (Fig. 1).
Mechanisms of SARS-CoV2-induced heart diseases
The potential mechanisms of SARS-CoV2 in the induction of heart disease are not clear. However, different pathways were defined for COVID-19 pathogenesis: (1) viremia and direct infection of lung and heart; (2) recruitment of the innate immune system by macrophages and cytokine storm; (3) adaptive immune system activation, and; (4) death or recovery (Maisch 2019). Therefore, direct and indirect mechanisms can be defined for the pathogenesis of SARS-CoV2. During SARS-CoV2 infection, the ACE2 receptor may cause a direct myocardial involvement and lead to myocarditis in severe patients (Turner et al. 2004; Zheng et al. 2020). In a recent study, ACE2 has been shown to express remarkably in heart tissue (Zou et al. 2020) and cardiac overexpression of ACE2 was demonstrated to protect the heart from ischemia-induced impairment (Der Sarkissian et al. 2008). The distribution of ACE2 in heart tissue, especially in pericytes, endothelial cells, cardiomyocytes, cardiofibroblasts, and epicardial adipose cells, and vascular system e.g., migratory angiogenic cells, smooth muscle cells, and endothelial cells increases the risk of direct infection of heart tissue (Gheblawi et al. 2020). SARS-CoV was reported to directly induce myocardial inflammation, and down-regulated myocardial ACE2 contributes to the myocardial abnormalities and cardiac consequences in SARS patients (Oudit et al. 2009b). In a murine model, it was confirmed that SARS-CoV causes an ACE2-dependent myocardial infection, as well as pulmonary infection (Oudit et al. 2009a). In the post-mortem evaluations of the cardiac tissue from a single patient, the viral RNA of SARS-CoV2 was detected, while histopathological evidence of myocarditis was not confirmed (Fan et al. 2020b). Thus, further investigations are still needed to confirm the direct-attach of virus and subsequent inflammation in the heart tissue, especially cardiac muscle, the leading cause of heart failure (Groß et al. 2020).
While the presence of ACE2 in cardiac tissue was not confirmed, the cytokine storm, a wide spectrum of infectious and non-infectious diseases, has been considered as the potential cause of heart injury (Wu et al. 2020c). The sources of cytokine storm are related to the secretion of inflammatory mediators from infected cells e.g., lung epithelial cells, dendritic cells, and macrophages (Gu et al. 2005). In COVID-19, apoptosis or necrosis of myocardial cells may result from the infiltration of inflammatory cells and the production of inflammatory mediators (Zhang et al. 2016), leading to the myocardial infarction (Xu et al. 2020b). The swelling of myocardial fibers and the presence of CD4+ T cells are the ultrastructural changes caused by viral infections (Huang et al. 2020; Warner et al. 2003). In another study, that the small arterioles, venules, and capillaries, containing plump endothelial cells, perivascular dying myocyte, and myocyte undergoing degeneration were detected in ultrastructural examinations. In the electron microscopy findings, the presence of SARS-CoV2 particles within the cardiac endothelial cell, but not present in the cardiac myocyte was confirmed. The higher infiltration of CD4+ and CD8+ lymphocytes were seen around the near the vascular structures and vascular endothelium, respectively (Fox et al. 2020). In SARS-CoV2-induced heart injury, the destruction of infected cells due to the stimulation of NLRP3 inflammasome, the main feature of the innate immune response to viral infection, leads to the production and systemic release of IL-1β (Sheng et al. 2020). The systemic inflammation can be associated with the progression of endothelial cell dysfunction and atherosclerosis, and increase the risk of cardiac ischaemia (Fatkhullina et al. 2016). Additionally, dysfunction of the coronary microvasculature due to increased levels of cytokines may result in myocardial injury (Libby 2020). The High levels of proinflammatory cytokines also contribute to the enhanced oxidative stress, reduced expression of endothelial nitric oxide synthase, and induced endothelial cell apoptosis (Kofler et al. 2005).
Due to the raised serum levels of proinflammatory mediators, like IL-1β, IL-6, IL-12, monocyte chemoattractant protein-1 (MCP-1), IFNγ, and IFN-inducible protein, SARS-CoV2 infection is related to the coagulation activation (Huang et al. 2020). In the pathological studies, megakaryocytes have been found in the microvasculature of the heart and bone marrow. These cells exert a critical role in diffuse microvascular thrombosis in COVID-19 (Rapkiewicz et al. 2020). Also, it has been observed that the COVID-19 is associated with the pathological features in the cardiovascular system, such as presence of inflammatory cells and apoptotic bodies in the cardiac tissue and acute myocyte necrosis. In the ultrastructural assessments, the viral inclusion bodies in vascular endothelial cells and CD61 + megakaryocytes were observed (Deshmukh et al. 2020). The histopathological findings of patients with a positive SARS-CoV2 result showed that the most common cardiac alterations were fibrosis in 14/14 (100%) and myocyte hypertrophy in 13/14 (93%) patients (Bradley et al. 2020). These findings confirm that the COVID-19-associated CVDs may be induced by the direct SARS-CoV2 infection and the indirect effects of infection including, cytokine storm, endothelial dysfunction, leucocytes infiltration, and formation of microvascular thrombosis.
COVID-19 and hematological abnormalities
Features of hematological abnormalities in COVID-19
SARS-CoV2 infection significantly affects the haematopoietic system and homeostasis is prominent (Debuc and Smadja 2020; Frater et al. 2020; Terpos et al. 2020). The most important laboratory finding with prognostic potential are lymphopenia (reduced peripheral CD4+ and CD8+ T cells) and leukopenia, which can increase the risk of bacterial infection (Tan et al. 2020a). The hematological abnormalities are more frequent in severe cases vs. mild cases; lymphocytopenia (< 1.0 × 109/L): 96.1% vs. 80.4%, leukopenia (WBC < 3700 cells/µL): 61.1% vs. 28.1%, and thrombocytopenia (platelet count < 100 × 109 cells per L): 57.7% vs. 31.6% (Mina et al. 2020). The results of a meta-analysis showed that thrombocytopenia is strongly correlated with the severity of illness (Lippi et al. 2020). Furthermore, peak platelet/lymphocyte ratio and neutrophil/lymphocyte ratio (NLR) are predictors of prognosis of COVID-19 (Terpos et al. 2020). The reduced absolute monocyte count and elevated NLR are associated with the higher rate of death mortality (Pakos et al. 2020). Furthermore, coagulation abnormalities e.g., severe thrombocytopenia, blood hypercoagulability, activated partial thromboplastin time, prolonged prothrombin time, and enhanced fibrin degradation products increase the risk of disseminated intravascular coagulation (DIC) among hospitalized patients (Becker 2020). Venous thromboembolism was reported in up to 10% for critically ill patients with COVID-19 (Moores et al. 2020). Anemia is not a frequent factor among COVID-19 patients, but the lower hemoglobin was detected in severe patients (Huang et al. 2020).
The surge in the symptoms of disease is correlated with enhanced concentrations of inflammatory mediators (e.g., TNF-α, IFNγ-induced protein 10 [IP-10], MCP-1, macrophage inflammatory proteins 1A [MIP1A], granulocyte colony-stimulating factor, ILs [2, 6, and 7], and the induction of cytokine storm and hyperinflammation syndrome (Lillicrap 2020; Quirch et al. 2020). Additionally, the inflammatory indices, such as including lactate dehydrogenase (LDH), CRP, and IL-6, help to detect the infected cases and improve the consequences of disease (Chen et al. 2020c; Li et al. 2020c; Ling 2020). CRP and NLR, the biomarkers of inflammation, are used for the diagnosis and assessment of many diseases. The CRP value above 100 mg/L is an indicator of severe bacterial infection (Cals et al. 2010; Gille-Johnson et al. 2012).
Higher levels of other biomarkers, such as serum D-dimer, ferritin, and procalcitonin are accompanied by poor prognosis (Huang et al. 2020c). Increased d-dimer may reflect the progress of DVT and/or pulmonary embolism, and can predict both severity and mortality of disease (Aggarwal et al. 2020). More recently, high serum concentrations of cortisol were reported to be attributed to the severity of disease and poor prognosis of treatment in critically ill COVID-19 patients (Tan et al. 2020b). Moreover, LDH is a non-specific indicator of different pathological conditions with cellular death and tissue damage (Wu et al. 2020f).
Mechanisms of SARS-CoV2-induced hematological abnormalities
Lymphopenia seems to be attributed to the ACE2 presence on the lymphocytes and direct infection of these cells (Wang et al. 2020e), whereas the elevated levels of cytokines may result in the lymphocyte apoptosis (Unsinger et al. 2009). Moreover, atrophy of lymphoid organs, including the spleen and lymph node showed the effects of cytokine storm, leading to further impairments of lymphocyte turnover (Cao 2020). The lung, a platelet biogenesis site, involves in hematopoiesis function and acts as a reservoir for megakaryocytes and haematopoietic progenitors (Lefrançais et al. 2017). Therefore, the platelet intake and/or its decreased production in damaged lungs of COVID-19 patients may result in thrombocytopenia (Liu et al. 2020c).
Coagulation abnormalities are multifactorial conditions, associate with the combination of inflammation, activation of platelet, endothelial dysfunction, immobility, and stasis of blood flow (The Lancet 2020). The higher levels of Factor VIII and von Willebrand factor proved the endothelial injury in COVID-19 patients (Escher et al. 2020). Endothelial cells may be affected by SARS-CoV2 due to the expression of ACE2, suggesting the direct infection of these cells in COVID-19 (Varga et al. 2020). Also, systemic inflammation may activate the endothelial cells via two pathways: increasing the interaction of cells with eukocyte and platelets and stimulating the expression of proinflammatory factors in endothelial cells, resulting in enhanced vascular permeability (Yang et al. 2016). It was revealed that endotheliopathy in COVID-19 was related to the severity of illness and death (Goshua et al. 2020). The higher CRD level is a diagnostic factor for pneumonia, correlated with the levels of inflammation. CRD stimulates the complement functions and enhance phagocytosis (Wang 2020). The activation of the complement system via SARS-COV2 in the lungs and other organs may are essential role in the initiation of acute and chronic inflammation, microthrombus formation, endothelial dysfunction, and DIC, and eventually induces multiorgan dysfunction and death (Noris et al. 2020). The high serum levels of C5a was confirmed in the infected cases, principally in critically ill cases (Gao et al. 2020). Serum ferritin is a biomarker for diagnosing the acute phase of inflammation and the macrophage activation syndrome (Kernan and Carcillo 2017).
COVID- 19 and renal diseases
Features of renal diseases in COVID-19
SARS-CoV-2 not only induces the diffused alveolar injury and acute respiratory failure, but also involves other organs, such as kidney and leads to renal resident cells injury (Huang et al. 2020). AKI is a common feature of renal disease, occurred in 5–15% of patients with SARS- and MERS-CoV infection with higher rate of mortality (Raza et al. 2020) In the first reports, the AKI incidence was few among COVID-19 patients. However, recent studies confirmed kidney failure in SARS COV2, related to the hospital death of cases with severe COVID-19 (Cheng et al. 2020a; Wang et al. 2020d). According to the data from hospitalized patients, the incidence of AKI was reported 29% among severe cases and reached to 69.57% in ≥ 60 year-old age group (Diao et al. 2020).
Various investigations confirmed that the AKI development significantly enhanced mortality among hospitalized COVID-19 patients (AKI 5.3 times vs. chronic diseases 1.5 times) (Li et al. 2020e; Rep 2020; Su et al. 2020a; Xu et al. 2020a). Also, the mortality rates increased significantly in hospitalized cases with proteinuria, higher baseline creatinine and BUN, hematuria, and AKI stage II and III (Cheng et al. 2020a).
In the previous study on 59 patients with COVID-19, the progressed albuminuria during the first days of admission was reported in 34% of patients, and 63% had proteinuria during their hospitalization (Li et al. 2020f). Increased blood urea nitrogen (BUN) level was also detected in 27% of all patients and two-third who died. Reduced kidney density in CT-scan implied on the tissue inflammation and edema (Boroujeni et al.).
Many patients with kidney injury such as abnormal urinary analysis and kidney dysfunction (increased BUN and serum creatinine) were detected during admission. Serum creatinine and BUN was increased in 15.5% and 14.1% of patients, respectively (Cheng et al. 2020c). In 138 COVID-19 patients with the ICU admission, the increased levels of creatine kinase was shown to be associated with kidney injury (Cheng et al. 2020c; Wang et al. 2020b). AKI was considered independently as a risk factor for patients’ mortality (Cheng et al. 2020c; Fan et al. 2020b). Patients with functional deficits in both innate and adaptive immunities are at higher risk for chronic kidney disease during the SARS-CoV2 pandemic (Cheng et al. 2020c) (Fig. 1).
Mechanisms of SARS-CoV2-induced kidney diseases
The signaling pathways involved in the kidney disease related to the COVID-19 is unknown, associated with several factors (Benedetti et al. 2020). Direct cellular infection though ACE2 or cytokine storm syndrome due to virus infection is supposed as a renal involvement mechanism (Naicker et al. 2020). ACE2 is expressed in different cells of kidney tissue e.g., proximal tubule epithelial cells, glomerular endothelial cells, and podocytes and kidney vasculature (Gheblawi et al. 2020). Key processes in human physiology are organized by an important signaling pathway of vasoactive peptides, called renin/Ang/aldosterone system. Interference of SARS-CoV and SARS-CoV2 with this system is performed via the ACE2 receptor (Ingraham et al. 2020). ACE2 with higher affinity SARS-CoV2 than SARS CoV (Wrapp et al. 2020), is expressed on various type renal cells, including brush border of proximal tubular cells, glomerular endothelial, mesangial cells, and podocytes (small quantity) (Ye et al. 2006). ACE2 also is expressed in the epithelium of glomerulus and smooth muscle of interlobular arteries (Zhang et al. 2004). Consuming of the inhibitors renin/Ang/aldosterone system and the blockers of Ang-receptor during COVID-19 may alter ACE2 expression and cause to increased mortality rate (Diaz 2020; Fang et al. 2020; Sommerstein and Gräni 2020; Vaduganathan et al. 2020).
In the renal samples from two COVID-19 patients with high-risk APOL1 genotype, the SARS-CoV2 RNA was not detected. The observed pathologic features were associated with collapsing focal segmental glomerulosclerosis. Also, the cytokine storm enhanced the gene expression of APOL1, resulting in podocyte damage (Kissling et al. 2020; Larsen et al. 2020). However, in the postmortem renal tissues from six COVID-19 patients with AKI exerted the pathologic features, such as severe acute tubular necrosis, as well as infiltration of macrophage and lymphocyte. Furthermore, the N-protein of SARS-CoV2 was identified in renal tubules and particles of CoV were identified in podocytes and tubular epithelium, suggesting that this virus can directly invade the renal cells (Abbate et al. 2020; Diao et al. 2020; Farkash et al. 2020; Su et al. 2020b).
The other indirect mechanisms which contribute to the renal injury may be related to hypoxia, hemodynamic instability and shock, sepsis, cytokine storm, and rhabdomyolysis (Cheng et al. 2020b; Kumar et al. 2009; Naicker et al. 2020). A recent study reported a decrease in kidney density using CT. Additionally, edema and inflammation of the renal parenchyma were identified (Li et al. 2020e). AKI development following COVID-19 could also be related to the activation of complement components, such as C5b-9. The strong deposition of C5b-9 was reported on the tubular cells than glomeruli and capillaries that resulted in parenchymal damages of renal tissue (Diao et al. 2020). The infiltration of inflammatory cells, e.g., CD4+ T cells, CD68+ macrophages, and CD56+ natural killer cells were observed in the tubules and interstitial tissue of patients (Diao et al. 2020). The intense activity of these immune cells may finally lead to apoptosis in epithelial cells, fibrosis, and alterations in the microvascular system (Saffarzadeh et al. 2012).
The involvement of microvascular thrombosis in renal samples of COVID-19 patients has not been certainly recognized. In the autopsy studies of renal tissue form COVID-19 patients, fibrin thrombi were rarely detected in glomerular regions related to the severe endothelial injury (Su et al. 2020b). The pathologic features of the kidney after COVID-19 may be associated with the direct invasion of virus, or the viral induced changes. The activation of complement seems and cytokine storm, infiltration of inflammatory cells, rarely thrombosis may mainly contribute to the renal damages, observed in the COVID-19 patients.
SARS-CoV2 and liver diseases
Features of liver diseases in COVID-19
It has been found that SARS-CoV2 is related to the dysfunction or damage of liver tissue, and after the lung, it seems to be the second organ (Xu et al. 2020b). Acute liver failure (ALF) in COVID-19 patients may result from the virus invasion, which directly infects liver cells. Some infections of the upper respiratory tract can influence the liver (Wu et al. 2020e). The function of the liver can be considered as a marker of disease progression, as the high frequency of serious COVID-19 cases showed of liver injury than mild cases (Zhang et al. 2020a). Several studies on COVID-19 patients primarily have demonstrated various degrees of raised serum biochemistries of the liver, such as irregular aspartate aminotransferase (AST), alanine aminotransferase (ALT), and total bilirubin (TB) levels (Garrido et al. 2020; Guan et al. 2020). In the evaluation of COVID-19 patients (n = 148), the rates of high liver biomarkers were estimated accordingly: LDH: 35.1%, AST:21.6%, ALT:18.2%, gamma-glutamyl transferase (GGT): 17.6%, TB: 6.1%, and Alkaline phosphatase (ALP):4.1% (Fan et al. 2020a). In another study, the elevated level of AST was identified in ICU admitted patients (62%) more frequently than other hospitalized patients (25%). Therefore, the ALF was predominant in critically ill cases than mild cases (Xu et al. 2020b). Elevated GGT, a diagnostic factor of cholangiocyte injury, and ALP levels were reported in 30/56 (54%) and 1/56 (1.8%), respectively among COVID-19 patients during hospitalization (Zhang et al. 2020b). The higher levels of ALF indicators, especially AST, are strongly attributed to the higher mortality risk (Lei et al. 2020).
In a preliminary observative study, the abdominal imaging of patients with COVID-19 was analyzed. In right upper quadrant ultrasounds, 20/37 (54%) showed a dilated sludge-filled gallbladder, suggesting the cholestasis in these patients. The fatty liver was also reported in 10/37 of patients (27%) and portal venous gas was confirmed by ultrasound and CT findings (Bhayana et al. 2020). Hypoalbuminaemia, caused by inadequate nutrition intake and overconsumption, has been observed in severe COVID-19 patients (Kim et al. 2017). The lower level of albumin in COVID-19 was reported to be the indicator of severe disease (Chen et al. 2020b). Furthermore, abnormal liver function was more likely to seen in infected patients with elevated levels of procalcitonin and CRP (Fan et al. 2020b) (Fig. 1).
Mechanisms of SARS-CoV2-induced liver diseases
Although the exact mechanisms of SARS-C0V-2 in the induction of ALF has not been identified, the main direct and indirect mechanism can be defined. Direct viral impact, systemic inflammation, drug-induced damage, congestion abnormalities, and hypoxia-induced damage are the factors, which contribute to liver damage in COVID-19 patients (Chau et al. 2004). The ACE2 receptors also are well presented in the liver cells (Qi et al. 2020; Zou et al. 2020), including cholangiocytes (60%) and hepatocytes (3%); and are absent in Kuppfer cells (Uhlén et al. 2015). The infection with SARS-CoV2 primarily is related to the invasion of the liver cells receptors to enter the objective cell. Diarrhoea was reported in about 2% to 10% of infected patients, and RNA of SARS-CoV2 was extracted from stool and blood samples, confirming the direct infection of host cells by the virus (Wang et al. 2020c). Bile duct cells with a critical role in liver recovery and immune reaction, express ACE2 higher than other liver cells, confirming that the infection of bile duct cells and not liver cells may involve the pathology of liver injury (Banales et al. 2019). Histopathological evaluation confirmed the low titer of SARS-CoV in the liver (Chau et al. 2004). However, viral inclusions were not observed in the liver samples from a case who died due to SARS-CoV2 infection (Xu et al. 2020b).
Microscopically, the liver manifests increased small lymphocytes infiltration in sinusoidal spaces, multifocal hepatic necrosis both in the periportal area and near the terminal hepatic veins, increased number of portal veins associated with luminal severe dilatation, and activated Kupffer cells with large cytoplasm containing necrotic debris (Cheng et al. 2020b; Naicker et al. 2020). The biomarkers of inflammation including CRP, serum ferritin were significantly raised in severe COVID-19 patients (Liu et al. 2020b). Furthermore, the inflammatory response with higher levels of ILs (1, 6 and 10) was prominent in SARS patients with ALF compared to normal liver function (Duan et al. 2003). In liver biopsies form SARS patients, the pathological features, such as eosinophilic bodies, mitotic cells, and balloon-like liver cells were observed, resulting in apoptosis and ALF (Chau et al. 2004). In MERS patients with liver damage, a mild inflammation of lobular lymphocytic and portal tract, as well as a mild hydropic degeneration of liver parenchymal cells were described (Alsaad et al. 2018; Ng et al. 2016). The post-mortem liver tissue analysis of COVID-19 patients presented only microvesicular steatosis, a common feature of sepsis (Koskinas et al. 2008).
The liver involvement in COVID-19 can be useful to divide into two groups; the first type of injury with mild liver abnormalities, related to a nonspecific reaction to the general inflammation (Li and Xiao 2020), and the second type of injury is related to with higher levels of TB, ALT, and AST which should be considered by clinicians (Li and Xiao 2020). Another manifestation of hepatic injury in COVID-19 patients is the fragmentation of the Smooth muscle layer of the portal vein (Wang et al. 2020c). In the similar respiratory viruses, the higher levels of liver biomarkers seem to be associated with liver injury due to interactions of Kupffer cells and cytotoxic T cells (Adams and Hubscher 2006). It believes that liver injury as a result of cytotoxic T cell activation and dysregulated innate immune responses are more potential to explain the correlation between raised liver biomarkers and COVID-19 severity (Bangash et al. 2020).
Furthermore, liver injury in severely infected patients was observed with relative decreased platelet counts, activation of fibrinolytic and coagulative signaling pathways, enhanced neutrophil counts and NLR, and high ferritin levels (Wang et al. 2020c). The immune deregulation and coagulation activation may alter the systemic metabolism of iron secondary to macrophage function (Sapey et al. 2019).
Additionally, drug-induced liver injury and abnormal liver blood test after therapeutics was observed in infected patients with SARS-CoV2, which should be considered by clinicians as a potential contributing factor. However, a mild liver test instability at baseline was recorded in many COVID-19 patients before treatment (Bangash et al. 2020). Hepatic injuries in COVID-19 patients may be caused by drugs like non‐steroidal anti‐inflammatory drugs and other herbal remedies (Li and Xiao 2020). According to the findings, the direct invasion of bile duct cells, imbalanced immune responses, coagulation abnormalities, systemic inflammation, and hypoxic condition are possible mechanisms of hepatic dysfunction in COVID-19 patients.
COVID-19 and Neurological diseases
Features of neurological diseases in COVID-19
Although the pulmonary disease following COVID-19 is the major leading cause of death (Wu et al. 2020a), the neurological manifestations need to be considered. In critically ill patients admitted to ICU, the incidence of neurological manifestation was reported higher than other patients. The neurological symptoms were characterized as the central nervous system (CNS) in severe cases and the peripheral nervous system (PNS) in mild to moderate cases (Mao et al. 2020). The PNS symptoms were hypoplasia, neuralgia, fatigue, myalgia, anosmia (or hyposmia), and hypogeusia (De Corato et al. 2011; Mao et al. 2020; Sun et al. 2020; Yang et al. 2020a). CNS manifestations, such as head pain, reduced consciousness, dizziness, ataxia, acute cerebrovascular disease, and epilepsy were recorded (Mao et al. 2020). In a report, the structural particles of SARS-CoV were detected in the brains of COVID-19 patients (Gu et al. 2005). Different neuropathologic conditions, such as acute flaccid paralysis (Turgay et al. 2015), encephalitis (Morfopoulou et al. 2016; Nilsson et al. 2020), and other neurological manifestations (Algahtani et al. 2016; Gu and Korteweg 2007; Li et al. 2016; Tsai et al. 2005) were reported in patients with HCoVs. In the previous study, the link between CoV and neurological disorders with demyelination, such as multiple sclerosis has been revealed (Khateb et al. 2020). In MRI of one case with SARS-CoV2 infection and seizures, demyelination lesions were observed in brain regions, including the periventricular white matter and the bulbo-medullary junction with the negative SARS-CoV2 RNA in CSF (Zanin et al. 2020). A patient with acute necrotizing encephalopathy (ANE) with positive CSF for RNA of SARS-CoV2 (Virhammar et al. 2020) and an 11-year-old pediatric case with encephalitis following COVID-19 (McAbee et al. 2020) were recorded. The incidence of brain stroke as a cerebrovascular complication was 5% among severe COVID-19 patients (Avula et al. 2020; Li et al. 2020d). Likewise, SARS-CoV2-induced encephalopathy has been confirmed in a case report (Malhotra 2016). The head CT images of a COID-19 patient (female) showed that bilateral medial thalami exerted symmetric hypoattenuation. In MRI images, hemorrhagic lesions were recorded in the medial temporal lobes, bilateral thalami, and subinsular areas (Poyiadji et al. 2020) (Fig. 1).
Mechanisms of SARS-CoV2-induced neurological diseases
The pathogenesis of SARS-CoV2 like other HCoVs in the nervous system also is related to (1) direct attach of nerve endings in PNS and transmission into the CNS (Rajčani 2003) or (2) indirect injuries related to the cytokine storm and viral sepsis (Li et al. 2020b). Increased inflammatory mediators in the nerve endings may induce the PNS manifestations (Ferrari et al. 2006). Also, the transmission of SARS-CoV2 through neural routes, such as olfactory nerve can increase the risk of infection in CNS, especially in the medulla oblongata, a regulator of cardiorespiratory system. The infection of this region may affect the spontaneous breath of COVID-19 patients and increase the mortality rate in ICU admitted patients (Kang et al. 2020). This virus may use a neuronal retrograde to infect the CNS, increasing the possibility of viral transmission via olfactory nerve endings in the nasal region (Hwang 2006; Netland et al. 2008; Xu et al. 2005). On the other hand, the distribution of ACE2 in the brainstem, mainly in the nuclei served as regulators of cardiorespiratory system, as well as other areas e.g., raphe and motor cortex has been identified (Doobay et al. 2007). Moreover, HCoVs may use a hematogenous route in which viral agent utilizes the bloodstream to infect CNS; infected leukocytes virus act as a vector or infected endothelial cells of blood-brain-barrier (BBB) with the ability of ACE2 expression may enhance the chance of CNS infection (Desforges et al. 2019; Rabelo et al. 2011). In the MRI study of COVID-19 case with CSF positive RNA of SARS-CoV2, symmetric pathologic signals in some brain regions, such as medial temporal lobes, subinsular region, central thalami, and brain stem. In the CSF, the high levels of tau and neurofilament light, the markers of neuronal injury glial fibrillary acidic protein, an astrocytic activation marker, as well as neuronal rescue proteins were observed, emphasizing the neurotropism of SARS-CoV2 (Virhammar et al. 2020). In histopathological evaluations, hemorrhagic lesions (foci of intraparenchymal blood with peripheral macrophages) in the white matter were observed. These lesions with the appearance of the ANE were associated with a wide range of alterations in the brain tissue, including axonal injuries, microscopic regions of necrosis, and microscopic infarcts, (Reichard et al. 2020).
The cytokine storm also has an essential role and contributes to the neurological manifestation of COVID-19 (Lei et al. 2020; Ruan et al. 2020). Indirectly, the cytokine storm is associated with BBB disruption, demyelination, and lead to ANE in viral infections (Mehta et al. 2020). Coagulation abnormalities due to high inflammatory responses were confirmed in COVID-19 patients with stroke (Li et al. 2020d; Sproston and Ashworth 2018). In addition, the advanced MicroCLOTS in the brain microvascular may lead to brain dysfunctions in infected patients (Ciceri et al. 2020). Consequently, direct infection of neural cells, cytokine storm, infiltration of inflammatory cells, disruption of BBB, endothelial dysfunction, coagulation abnormalities, and hypoxia may have a critical role in the neurological manifestations of patients.
Conclusions
COVID-19 mainly in severe cases in addition to lung involves different organs such as heart, liver, and kidney, as well as hematological and nervous system, and induce multi-organ failure. SARS-COV2 may directly invade the host cells of different organs through the ACE2 receptor due to the presence of this receptor in these organs. On the other hand, activation of the complement system, cytokine storm, dysregulated immune responses, coagulation dysfunction, and infiltration of inflammatory cells in SARS-CoV2 infection can induce the multi-organ failure in these patients. Overall, understanding the clinical, laboratory and radiological features of COVID-19 in critically ill patients with multi-organ dysfunction should be clarified for clinicians. Consequently, increasing the knowledge on the pathophysiology of SARS-COV2-induced multi-organ failure may ultimately result in better ways to treat COVID-19 patients and decrease the associated morbidity and mortality.
References
Abbate M, Rottoli D, Gianatti A (2020) COVID-19 attacks the kidney: ultrastructural evidence for the presence of virus in the glomerular epithelium. Nephron Clin Pract 58:515–523
Ackermann M et al (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19 N. Engl J Med 383:120–128. https://doi.org/10.1056/NEJMoa2015432
Adams DH, Hubscher SG (2006) Systemic viral infections and collateral damage in the liver. Am J Pathol 168:1057–1059. https://doi.org/10.2353/ajpath.2006.051296
Aggarwal M, Dass J, Mahapatra M (2020) Hemostatic abnormalities in COVID-19: an update. Indian J Hematol Blood Transf. https://doi.org/10.1007/s12288-020-01328-2
Alberici F et al (2020) Management Of patients on dialysis and with kidney transplant during SARS-COV-2 (COVID-19) pandemic In Brescia, Italy Kidney International Reports
Algahtani H, Subahi A, Shirah B (2016) Neurological complications of middle east respiratory syndrome coronavirus: a report of two cases and review of the literature case. Rep Neurol Med 2016:3502683. https://doi.org/10.1155/2016/3502683
Alhogbani T (2016) Acute myocarditis associated with novel Middle East respiratory syndrome coronavirus. Ann Saudi Med 36:78–80
Alsaad KO et al (2018) Histopathology of Middle East respiratory syndrome coronovirus (MERS-CoV) infection–clinicopathological and ultrastructural study. Histopathology 72:516–524
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26:450
Avula A et al (2020) COVID-19 presenting as stroke. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2020.04.077
Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ (2019) Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol 16:269–281
Bangash MN, Patel J, Parekh D (2020) COVID-19 and the liver: little cause for concern. Lancet Gastroenterol Hepatol 5:529–530. https://doi.org/10.1016/S2468-1253(20)30084-4
Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S (2020) COVID-19 Autopsies, Oklahoma, USA . Am J Clin Pathol 153:725–733. https://doi.org/10.1093/ajcp/aqaa062
Baud D, Qi X, Nielsen-Saines K, Musso D, Pomar L, Favre G (2020) Real estimates of mortality following COVID-19 infection. Lancet Infect Dis. https://doi.org/10.1016/s1473-3099(20)30195-x
Becker RC (2020) COVID-19 update: Covid-19-associated coagulopathy. J Thromb Thrombolysis 50:54–67. https://doi.org/10.1007/s11239-020-02134-3
Benedetti C, Waldman M, Zaza G, Riella LV, Cravedi P (2020) COVID-19 and the kidneys: an update. Front Med. https://doi.org/10.3389/fmed.2020.00423
Bernstein KE, Khan Z, Giani JF, Cao DY, Bernstein EA, Shen XZ (2018) Angiotensin-converting enzyme in innate and adaptive immunity. Nat Rev Nephrol 14:325–336. https://doi.org/10.1038/nrneph.2018.15
Bhatia M, Zemans RL, Jeyaseelan S (2012) Role of chemokines in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol 46:566–572. https://doi.org/10.1165/rcmb.2011-0392TR
Bhayana R et al (2020) Abdominal imaging findings in COVID-19: preliminary observations. Radiology. https://doi.org/10.1148/radiol.2020201908
Boroujeni EK, Kellner SJ, Pezeshgi A (2021) Covid-19 and kidney; a mini-review on current concepts and new data. J Nephropharmacol 10:e01
Bradley BT et al (2020) Histopathology and ultrastructural findings of fatal COVID-19 infections in Washington State: a case series. Lancet. https://doi.org/10.1016/S0140-6736(20)31305-2
Burchfield J (2020) Renin-Angiotensin-Aldosterone system: double-edged sword in COVID-19 infection
Cabeça TK, Granato C, Bellei N (2013) Epidemiological and clinical features of human coronavirus infections among different subsets of patients. Influenza Other Respir Viruses 7:1040–1047. https://doi.org/10.1111/irv.12101
Cals JW, Schot MJ, de Jong SA, Dinant G-J, Hopstaken RM (2010) Point-of-care C-reactive protein testing and antibiotic prescribing for respiratory tract infections: a randomized controlled trial. Ann Family Med 8:124–133
Cao X (2020) COVID-19: immunopathology and its implications for therapy. Nat Rev Immunol 20:269–270. https://doi.org/10.1038/s41577-020-0308-3
Channappanavar R, Perlman S (2017) Pathogenic human coronavirus infections: causes and consequences of cytokine storm and immunopathology. Semin Immunopathol 39:529–539. https://doi.org/10.1007/s00281-017-0629-x
Chau TN et al (2004) SARS-associated viral hepatitis caused by a novel coronavirus: report of three cases . Hepatology (Baltimore Md) 39:302–310. https://doi.org/10.1002/hep.20111
Chen J et al (2020a) Clinical progression of patients with COVID-19 in Shanghai, China. J Infect 80:e1–e6
Chen N et al (2020b) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study . Lancet (London, England) 395:507–513. https://doi.org/10.1016/s0140-6736(20)30211-7
Chen X et al (2020c) Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely correlated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa449
Cheng Y et al (2020a) Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Intl
Cheng Y et al (2020b) Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney international 97:829–838. https://doi.org/10.1016/j.kint.2020.03.005
Cheng Y et al (2020c) Kidney impairment is associated with in-hospital death of COVID-19 patients medRxiv
Chung M et al (2020) CT imaging features of 2019 novel coronavirus (2019-nCoV). Radiology 295:202–207
Ciceri F et al (2020) Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resuscitation 22:95–97
Clerkin KJ et al (2020) Coronavirus disease 2019 (COVID-19) and cardiovascular disease. Circulation 141:1648
De Corato A, Lisi L, Capuano A, Tringali G, Tramutola A, Navarra P, Dello Russo C (2011) Trigeminal satellite cells express functional calcitonin gene-related peptide receptors, whose activation enhances interleukin-1β pro-inflammatory effects. J Neuroimmunol 237:39–46. https://doi.org/10.1016/j.jneuroim.2011.05.013
De Wit E, Van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523
Debuc B, Smadja DM (2020) Is COVID-19 a new hematologic disease? Stem Cell Rev Rep. https://doi.org/10.1007/s12015-020-09987-4
Der Sarkissian S, Grobe JL, Yuan L, Narielwala DR, Walter GA, Katovich MJ, Raizada MK (2008) Cardiac overexpression of angiotensin converting enzyme 2 protects the heart from ischemia-induced pathophysiology. Hypertension 51:712–718
Desforges M, Le Coupanec A, Dubeau P, Bourgouin A, Lajoie L, Dubé M, Talbot PJ (2019) Human coronaviruses and other respiratory viruses: underestimated opportunistic pathogens of the central nervous system? Viruses. https://doi.org/10.3390/v12010014
Deshmukh V, Motwani R, Kumar A, Kumari C, Raza K (2020) Histopathological observations in COVID-19: a systematic review. J Clin Pathol. https://doi.org/10.1136/jclinpath-2020-206995
Diao B et al (2020) Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection MedRxiv
Diaz JH (2020) Hypothesis: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J Travel Med. https://doi.org/10.1093/jtm/taaa041
Doobay MF, Talman LS, Obr TD, Tian X, Davisson RL, Lazartigues E (2007) Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am J Physiol Regul Integr Comp Physiol 292:R373–R381. https://doi.org/10.1152/ajpregu.00292.2006
Duan Z, Chen Y, Zhang J, Zhao J, Lang Z, Meng F, Bao X (2003) Clinical characteristics and mechanism of liver injury in patients with severe acute respiratory syndrome Zhonghua gan zang bing za zhi = Zhonghua ganzangbing zazhi = Chinese. J Hepatol 11:493–496
Escher R, Breakey N, Lämmle B (2020) Severe COVID-19 infection associated with endothelial activation. Thromb Res 190:62. https://doi.org/10.1016/j.thromres.2020.04.014
Fan Z et al (2020a) Clinical features of COVID-19-related liver damage. Clin Gastroenterol Hepatol. https://doi.org/10.1101/2020.02.26.20026971
Fan Z et al (2020b) Clinical features of COVID-19-related liver functional abnormality clinical. Gastroenterol Hepatol 18:1561–1566. https://doi.org/10.1016/j.cgh.2020.04.002
Fang L, Karakiulakis G, Roth M (2020) Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir Med 8:e21
Farkash EA, Wilson AM, Jentzen JM (2020) Ultrastructural evidence for direct renal infection with SARS-CoV-2. J Am Soc Nephrol 31:1683
Fatkhullina AR, Peshkova IO, Koltsova EK (2016) The role of cytokines in the development of atherosclerosis. Biochemistry (Mosc) 81:1358–1370. https://doi.org/10.1134/S0006297916110134
Fauci AS, Lane HC, Redfield RR (2020) Covid-19: navigating the uncharted N. Engl J Med 382:1268–1269. https://doi.org/10.1056/NEJMe2002387
Ferrari S, Vento S, Monaco S, Cavallaro T, Cainelli F, Rizzuto N, Temesgen Z Human immunodeficiency virus-associated peripheral neuropathies. In: Mayo Clinic Proceedings (2006) vol 2. Elsevier, pp 213–219
Fox SE, Li G, Akmatbekov A, Harbert JL, Lameira FS, Brown JQ, Vander Heide RS (2020) Unexpected features of cardiac pathology in COVID-19 infection. Circulation. https://doi.org/10.1161/circulationaha.120.049465
Frater JL, Zini G, d’Onofrio G, Rogers HJ (2020) COVID-19 and the clinical hematology laboratory. Intl J Lab Hematol 1:11–18
Gao T et al (2020) Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation MedRxiv
Garrido I, Liberal R, Macedo G (2020) COVID-19 and liver disease‐what we know on 1st May 2020. Aliment Pharmacol Therap
Gheblawi M et al (2020) Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the Renin-Angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res 126:1456–1474. https://doi.org/10.1161/CIRCRESAHA.120.317015
Gille-Johnson P, Hansson KE, Gårdlund B (2012) Clinical and laboratory variables identifying bacterial infection and bacteraemia in the emergency department . Scan J Infect Dis 44:745–752
Goshua G et al (2020) Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 7:e575–e582. https://doi.org/10.1016/s2352-3026(20)30216-7
Grillet F, Behr J, Calame P, Aubry S, Delabrousse E (2020) Acute pulmonary embolism associated with COVID-19 pneumonia detected with pulmonary. CT Angiogr Radiol 296:E186–E188. https://doi.org/10.1148/radiol.2020201544
Groß S, Jahn C, Cushman S, Bär C, Thum T (2020) SARS-CoV-2 receptor ACE2-dependent implications on the cardiovascular system: from basic science to clinical implications. J Mol Cell Cardiol 144:47–53. https://doi.org/10.1016/j.yjmcc.2020.04.031
Gu J, Korteweg C (2007) Pathology and pathogenesis of severe acute respiratory syndrome. Am J Pathol 170:1136–1147. https://doi.org/10.2353/ajpath.2007.061088
Gu J et al (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202:415–424. https://doi.org/10.1084/jem.20050828
Guan W-j et al (2020) Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med 382:1708–1720
Gupta A et al (2020) Extrapulmonary manifestations of COVID-19. Nat Med 26:1017–1032. https://doi.org/10.1038/s41591-020-0968-3
Guzik TJ et al (2020) COVID-19 and the cardiovascular system: implications for risk assessment, diagnosis, and treatment options. Cardiovasc Res 116:1666–1687. https://doi.org/10.1093/cvr/cvaa106
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637. https://doi.org/10.1002/path.1570
Hładij R, Rajtar-Salwa R, Dimitrow PP (2017) Troponin as ischemic biomarker is related with all three echocardiographic risk factors for sudden death in hypertrophic cardiomyopathy (ESC Guidelines 2014). Cardiovasc Ultrasound 15:24
Huang C et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395:497–506
Huang I, Pranata R, Lim MA, Oehadian A, Alisjahbana B (2020c) C-reactive protein, procalcitonin, D-dimer, and ferritin in severe coronavirus disease-2019: a meta-analysis. Therap Adv Respir Dis 14:1753466620937175
Hwang CS (2006) Olfactory neuropathy in severe acute respiratory syndrome: report of A case. Acta Neurol Taiwan 15:26–28
Imai Y et al (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116. https://doi.org/10.1038/nature03712
Ingraham NE et al (2020) Understanding the renin-angiotensin-aldosterone-SARS-CoV-axis: a comprehensive review. Eur Respir J 56:2000912
Jiang F, Deng L, Zhang L, Cai Y, Cheung CW, Xia Z (2020) Review of the clinical characteristics of coronavirus disease 2019 (COVID-19). J General Inter Med 35:1545
Jin YH et al (2020) A rapid advice guideline for the diagnosis and treatment of 2019 novel coronavirus (2019-nCoV) infected pneumonia (standard version). Mil Med Res 7:4. https://doi.org/10.1186/s40779-020-0233-6
Kang S et al (2020) Recent progress in understanding 2019 novel coronavirus (SARS-CoV-2) associated with human respiratory disease: detection, mechanisms and treatment. Int J Antimicrob Agents. https://doi.org/10.1016/j.ijantimicag.2020.105950
Kemp M, Donovan J, Higham H, Hooper J (2004) Biochemical markers of myocardial injury. Br J Anaesth 93:63–73
Kernan KF, Carcillo JA (2017) Hyperferritinemia inflammation. Int Immunol 29:401–409. https://doi.org/10.1093/intimm/dxx031
Khateb M, Bosak N, Muqary M (2020) Coronaviruses and central nervous system manifestations. Front Neurol 11:715. https://doi.org/10.3389/fneur.2020.00715
Kim S, McClave SA, Martindale RG, Miller KR, Hurt RT (2017) Hypoalbuminemia and clinical outcomes: what is the mechanism behind the relationship? Am Surg 83:1220–1227
Kissling S et al (2020) Collapsing glomerulopathy in a COVID-19 patient. Kidney Intl 98:228–231. https://doi.org/10.1016/j.kint.2020.04.006
Kofler S, Nickel T, Weis M (2005) Role of cytokines in cardiovascular diseases: a focus on endothelial responses to inflammation. Clin Sci (London, England: 1979) 108:205–213. https://doi.org/10.1042/cs20040174
Koskinas J et al (2008) Liver histology in ICU patients dying from sepsis: a clinico-pathological study. World J Gastroenterol 14:1389–1393. https://doi.org/10.3748/wjg.14.1389
Kumar A et al (2009) Critically ill patients with 2009 influenza A (H1N1) infection in Canada. JAMA 302:1872–1879
Larsen CP, Bourne TD, Wilson JD, Saqqa O, Sharshir MA (2020) Collapsing glomerulopathy in a patient with COVID-19. Kidney Intl Rep 5:935–939. https://doi.org/10.1016/j.ekir.2020.04.002
Lee YB, Nagai A, Kim SU (2002) Cytokines, chemokines, and cytokine receptors in human microglia. J Neurosci Res 69:94–103. https://doi.org/10.1002/jnr.10253
Lefrançais E et al (2017) The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544:105–109
Lei F et al (2020) Longitudinal association between markers of liver injury and mortality in COVID-19 in China . Hepatology 72:389–398. https://doi.org/10.1002/hep.31301
Li F (2016) Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 3:237–261. https://doi.org/10.1146/annurev-virology-110615-042301
Li X, Ma X (2020) Acute respiratory failure in COVID-19: is it “typical” ARDS? Crit Care 24:1–5
Li Y, Xiao SY (2020) Hepatic involvement in COVID-19 patients: pathology, pathogenesis and clinical implications. J Med Virol. https://doi.org/10.1002/jmv.25973
Li Y et al (2016) Coronavirus infections in the central nervous system and respiratory tract show distinct features in hospitalized children. Intervirology 59:163–169. https://doi.org/10.1159/000453066
Li G et al (2020a) Coronavirus infections and immune responses. J Med Virol 92:424–432. https://doi.org/10.1002/jmv.25685
Li H et al (2020b) SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. https://doi.org/10.1016/s0140-6736(20)30920-x
Li X et al (2020c) Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol 146:110
Li Y et al (2020d) Acute cerebrovascular disease following COVID-19: a single center, retrospective, observational study. Stroke Vasc Neurol. https://doi.org/10.1136/svn-2020-000431
Li Z et al (2020e) Caution on kidney dysfunctions of COVID-19 patients
Li Z et al (2020f) Caution on kidney dysfunctions of COVID-19 patients. MedRxiv
Libby P (2020) The heart in COVID-19: primary target or secondary bystander? JACC Basic Transl Sci 5:537–542. https://doi.org/10.1016/j.jacbts.2020.04.001
Lillicrap D (2020) Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. JTH 18:786–787. https://doi.org/10.1111/jth.14781
Lim YX, Ng YL, Tam JP, Liu DX (2016) Human coronaviruses: a review of virus-host. Interact Dis 4:26. https://doi.org/10.3390/diseases4030026
Ling W (2020) C-reactive protein levels in the early stage of COVID-19. Med Mal Infect 50:332
Lippi G, Plebani M, Henry BM (2020) Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a meta-analysis. Clin Chim Acta Intl J Clin Chem 506:145–148. https://doi.org/10.1016/j.cca.2020.03.022
Liu X, Zhang R, He G (2020c) Hematological findings in coronavirus disease 2019: indications of progression of disease Annals of hematology 99:1421-1428 doi:10.1007/s00277-020-04103-5
Liu DX, Liang JQ, Fung TS (2020a) Human Coronavirus-229E, -OC43, -NL63, and -HKU1 reference module in life sciences: B978-970-912-809633-809638.https://doi.org/10.1016/B978-0-12-809633-8.21501-X
Liu J et al (2020b) Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 55:102763
Liu Z et al (2020d) Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J Med Virol. https://doi.org/10.1002/jmv.25726
Lu R et al (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding . Lancet (London England) 395:565–574. https://doi.org/10.1016/s0140-6736(20)30251-8
Maisch B (2019) Cardio-immunology of myocarditis: focus on immune mechanisms and treatment options. Front Cardiovasc Med 6:48. https://doi.org/10.3389/fcvm.2019.00048
Malhotra R (2016) Understanding migraine: potential role of neurogenic inflammation. Ann Indian Acad Neurol 19:175
Mao L et al (2020) Neurological manifestations of hospitalized patients with COVID-19 in Wuhan. a retrospective case series study, China
McAbee GN, Brosgol Y, Pavlakis S, Agha R, Gaffoor M (2020) Encephalitis associated with COVID-19 infection in an 11-year-old. Child Pediatr Neurol 109:94. https://doi.org/10.1016/j.pediatrneurol.2020.04.013
Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, Collaboration HAS (2020) COVID-19: consider cytokine storm syndromes and immunosuppression . Lancet (London England) 395:1033
Mina A, van Besien K, Platanias LC (2020) Hematological manifestations of COVID-19. Leukemia Lymphoma. https://doi.org/10.1080/10428194.2020.1788017
Moores LK et al (2020) Prevention, diagnosis, and treatment of VTE in patients with coronavirus disease 2019: CHEST guideline and expert panel Report chest:S0012-3692(0020)31625–31621. https://doi.org/10.1016/j.chest.2020.05.559
Morfopoulou S et al (2016) Human coronavirus OC43 associated with fatal encephalitis. N Engl J Med 375:497–498. https://doi.org/10.1056/NEJMc1509458
Mukherjee A, Ahmad M, Frenia D (2020) A coronavirus disease 2019 (COVID-19) patient with multifocal pneumonia treated with hydroxychloroquine. Cureus 12:e7473–e7473. https://doi.org/10.7759/cureus.7473
Naicker S, Yang CW, Hwang SJ, Liu BC, Chen JH, Jha V (2020) The novel coronavirus 2019 epidemic and kidneys. Kid Intl 97:824–828. https://doi.org/10.1016/j.kint.2020.03.001
Nainu F, Shiratsuchi A, Nakanishi Y (2017) Induction of apoptosis and subsequent phagocytosis of virus-infected cells as an antiviral. Mech Front Immunol 8:1220–1220. https://doi.org/10.3389/fimmu.2017.01220
Naqvi AAT et al (2020) Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta Mol Basis Dis 1866:165878–165878. https://doi.org/10.1016/j.bbadis.2020.165878
Ndrepepa G, Kastrati A (2018) Creatine kinase myocardial band-a biomarker to assess prognostically relevant periprocedural myocardial infarction. Int J Cardiol 270:118–119
Netland J, Meyerholz DK, Moore S, Cassell M, Perlman S (2008) Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE. 2 J Virol 82:7264–7275. https://doi.org/10.1128/jvi.00737-08
Neumann JT et al (2017) Early diagnosis of acute myocardial infarction using high-sensitivity troponin I. PLoS ONE. https://doi.org/10.1371/journal.pone.0174288
Ng DL et al (2016) Clinicopathologic, immunohistochemical, and ultrastructural findings of a fatal case of Middle East respiratory syndrome coronavirus infection in the United Arab Emirates, April 2014. Am J Pathol 186:652–658
Nilsson A, Edner N, Albert J, Ternhag A (2020) Fatal encephalitis associated with coronavirus OC43 in an immunocompromised. Child Infect Dis (Lond). https://doi.org/10.1080/23744235.2020.1729403
Noris M, Benigni A, Remuzzi G (2020) The case of complement activation in COVID-19 multiorgan impact. Kidney Intl 98:314–322. https://doi.org/10.1016/j.kint.2020.05.013
Oudit G, Kassiri Z, Jiang C, Liu P, Poutanen S, Penninger J, Butany J (2009a) SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with. SARS Eur J Clin Investig 39:618–625
Oudit GY, Kassiri Z, Jiang C, Liu PP, Poutanen SM, Penninger JM, Butany J (2009b) SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest 39:618–625. https://doi.org/10.1111/j.1365-2362.2009.02153.x
Pakos I et al (2020) Characteristics of peripheral blood differential counts in hospitalized patients with COVID-19. Eur J Haematol
Perlman S, Netland J (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7:439–450
Pollack A, Kontorovich AR, Fuster V, Dec GW (2015) Viral myocarditis—diagnosis, treatment options, and current controversies. Nat Rev Cardiol 12:670
Poyiadji N, Shahin G, Noujaim D, Stone M, Patel S, Griffith B (2020) COVID-19–associated acute hemorrhagic necrotizing encephalopathy: CT and MRI features Radiology:201187
Pyrc K, Berkhout B, van der Hoek L (2007) The novel human coronaviruses NL63 and HKU1. J Virol 81:3051–3057. https://doi.org/10.1128/jvi.01466-06
Qi F, Qian S, Zhang S, Zhang Z (2020) Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses Biochemical and biophysical research communications
Quirch M, Lee J, Rehman S (2020) Hazards of the cytokine storm and cytokine-targeted therapy in patients with COVID-19. J Med Intl Res 22:e20193
Rabelo LA, Alenina N, Bader M (2011) ACE2-angiotensin-(1–7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens Res 34:154–160. https://doi.org/10.1038/hr.2010.235
Rajčani J (2003) Molecular mechanisms of virus spread and virion components as tools of virulence . Acta Microbiologica et Immunologica Hungarica 50:407–431
Rapkiewicz AV et al (2020) Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: a case series. EClinicalMedicine 24:100434
Raza A, Estepa A, Chan V, Jafar MS (2020) Acute renal failure in critically Ill COVID-19 patients with a focus on the role of renal replacement therapy: a review of what we know. So Far Cureus 12:e8429–e8429. https://doi.org/10.7759/cureus.8429
Reichard RR, Kashani KB, Boire NA, Constantopoulos E, Guo Y, Lucchinetti CF (2020) Neuropathology of COVID-19: a spectrum of vascular and acute disseminated encephalomyelitis (ADEM)-like pathology. Acta Neuropathol 140:1–6. https://doi.org/10.1007/s00401-020-02166-2
Rep KI (2020) Management of Patients on Dialysis and With Kidney Transplantation Kidney International
Robles A et al (2011) Viral vs bacterial community-acquired pneumonia: Radiologic features. Eur Respiratory Soc
Rothan HA, Byrareddy SN (2020) The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak Journal of autoimmunity:102433
Ruan Q, Yang K, Wang W, Jiang L, Song J (2020) Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med 20:1–3. https://doi.org/10.1007/s00134-020-05991-x
Saffarzadeh M et al (2012) Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE 7:e32366
Sagar S, Liu PP, Cooper LT Jr (2012) Myocarditis. Lancet 379:738–747
Sahu KK, Mishra AK, Lal A (2020a) Comprehensive update on current outbreak of novel coronavirus infection (2019-nCoV). Ann Transl Med 8:393–393. https://doi.org/10.21037/atm.2020.02.92
Sahu KK, Mishra AK, Martin K, Chastain I (2020b) COVID-19 and clinical mimics. Correct diagnosis is the key to appropriate therapy Monaldi archives for chest disease = Archivio Monaldi per le malattie del torace 90 https://doi.org/10.4081/monaldi.2020.1327
Sapey E et al (2019) Simvastatin improves neutrophil function and clinical outcomes in pneumonia. A pilot randomized controlled clinical trial. Am J Respir Crit Care Med 200:1282–1293. https://doi.org/10.1164/rccm.201812-2328OC
Sheng CC et al (2020) Canakinumab to reduce deterioration of cardiac and respiratory function in SARS-CoV‐2 associated myocardial injury with heightened inflammation (canakinumab in Covid‐19 cardiac injury: The three C study. Clin Cardiol
Shi H, Han X, Zheng C (2020a) Evolution of CT manifestations in a patient recovered from 2019 novel coronavirus (2019-nCoV) pneumonia in Wuhan, China. Radiology 295:20. https://doi.org/10.1148/radiol.2020200269
Shi S et al (2020b) Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China JAMA cardiology
Sommerstein R, Gräni C (2020) Rapid response: preventing a COVID-19 pandemic: ACE inhibitors as a potential risk factor for fatal COVID-19. BMJ 368:m810
Sproston NR, Ashworth JJ (2018) Role of C-Reactive Protein at Sites of Inflammation and Infection. Front Immunol 9:754–754. https://doi.org/10.3389/fimmu.2018.00754
Su H et al (2020a) Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China Kidney international
Su H et al (2020b) Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in. China Kidney International 98:219–227. https://doi.org/10.1016/j.kint.2020.04.003
Sun P, Lu X, Xu C, Sun W, Pan B (2020) Understanding of COVID-19 based on current evidence. J Med Virol. https://doi.org/10.1002/jmv.25722
Tan L et al (2020a) Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal transduction targeted therapy 5:1–3
Tan T et al (2020b) Association between high serum total cortisol concentrations and mortality from COVID-19. Lancet Diabetes Endocrinol 8:659–660. https://doi.org/10.1016/s2213-8587(20)30216-3
Ternant D et al (2017) Revisiting myocardial necrosis biomarkers: assessment of the effect of conditioning therapies on infarct size by kinetic modelling. Sci Rep 7:1–12
Terpos E et al (2020) Hematological findings and complications of COVID-19 American journal of hematology
The Lancet H (2020) COVID-19 coagulopathy: an evolving story. The Lancet Haematology 7:e425. https://doi.org/10.1016/s2352-3026(20)30151-4
Tsai LK, Hsieh ST, Chang YC (2005) Neurological manifestations in severe acute respiratory syndrome. Acta Neurol Taiwan 14:113–119
Turgay C, Emine T, Ozlem K, Muhammet SP, Haydar AT (2015) A rare cause of acute flaccid paralysis: Human coronaviruses. J Pediatr Neurosci 10:280–281. https://doi.org/10.4103/1817-1745.165716
Turner AJ, Hiscox JA, Hooper NM (2004) ACE2: from vasopeptidase to SARS virus receptor. Trends Pharmacol Sci 25:291–294
Uhlén M et al (2015) Tissue-based map of the human proteome. Science 347:1260419
Unsinger J, McDonough JS, Shultz LD, Ferguson TA, Hotchkiss RS (2009) Sepsis-induced human lymphocyte apoptosis and cytokine production in “humanized" mice. J Leukoc Biol 86:219–227. https://doi.org/10.1189/jlb.1008615
Vaduganathan M, Vardeny O, Michel T, McMurray JJ, Pfeffer MA, Solomon SD (2020) Renin–Angiotensin–Aldosterone System Inhibitors in Patients with Covid-19 New England Journal of Medicine
Van Doremalen N et al (2020) Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1 . N Engl J Med 382:1564–1567
Varga Z et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418. https://doi.org/10.1016/s0140-6736(20)30937-5
Vasudevan G, Mercer DW, Varat MA (1978) Lactic dehydrogenase isoenzyme determination in the diagnosis of acute myocardial infarction. Circulation 57:1055–1057
Verdecchia P, Cavallini C, Spanevello A, Angeli F (2020) The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med 76:14–20. https://doi.org/10.1016/j.ejim.2020.04.037
Virhammar J et al (2020) Acute necrotizing encephalopathy with SARS-CoV-2 RNA confirmed in cerebrospinal fluid. Neurology 95:445–449. https://doi.org/10.1212/wnl.0000000000010250
Wang L (2020) C-reactive protein levels in the early stage of COVID-19. Médecine et Maladies Infectieuses 50:332–334. https://doi.org/10.1016/j.medmal.2020.03.007
Wang D et al (2020a) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. JAMA 323:1061–1069
Wang D et al (2020b) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China Jama
Wang D et al (2020c) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan. China Jama 323:1061–1069. https://doi.org/10.1001/jama.2020.1585
Wang D et al (2020d) Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. https://doi.org/10.1001/jama.2020.1585
Wang X et al (2020e) SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion Cellular & molecular immunology:1–3
Warner FJ, Guy JL, Lambert DW, Hooper NM, Turner AJ (2003) Angiotensin converting enzyme-2 (ACE2) and its possible roles in hypertension, diabetes and cardiac function. Lett Pept Sci 10:377–385
Wölfel R et al (2020) Virological assessment of hospitalized patients with COVID-2019. Nature 581:465–469. https://doi.org/10.1038/s41586-020-2196-x
Wrapp D et al (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion. conformation Science 367:1260–1263
Wu C et al (2020a) Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan. China JAMA Intern Med. https://doi.org/10.1001/jamainternmed.2020.0994
Wu C et al (2020b) Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China JAMA internal medicine
Wu C et al (2020c) Heart injury signs are associated with higher and earlier mortality in coronavirus disease 2019 (COVID-19) medRxiv
Wu J, Deng W, Li S, Yang X (2020d) Advances in research on ACE2 as a receptor for 2019-nCoV Cell Mol Life Sci:1–14 https://doi.org/10.1007/s00018-020-03611-x
Wu J, Song S, Cao H-C, Li L-J (2020e) Liver diseases in COVID-19: Etiology, treatment and prognosis World. J Gastroenterol 26:2286
Wu MY, Yao L, Wang Y, Zhu XY, Wang XF, Tang PJ, Chen C (2020f) Clinical evaluation of potential usefulness of serum lactate dehydrogenase (LDH) in 2019 novel coronavirus (COVID-19) pneumonia Respiratory research 21:171 https://doi.org/10.1186/s12931-020-01427-8
Xu J et al (2005) Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin Infect Dis 41:1089–1096. https://doi.org/10.1086/444461
Xu S et al (2020a) Acute kidney injury at early stage as a negative prognostic indicator of patients with COVID-19: a hospital-based retrospective analysis medRxiv
Xu Z et al (2020b) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. The Lancet Respiratory medicine 8:420–422. https://doi.org/10.1016/s2213-2600(20)30076-x
Xu H et al (2020c) Clinical characteristics and risk factors of cardiac involvement in COVID-19. J Am Heart Assoc 9:e016807
Yamamoto S et al (2011) Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis Arterioscler. Thromb Vasc Biol 31:2856–2864. https://doi.org/10.1161/atvbaha.111.237198
Yang X, Chang Y, Wei W (2016) Endothelial Dysfunction and Inflammation: Immunity in Rheumatoid Arthritis Mediators of inflammation 2016:6813016 https://doi.org/10.1155/2016/6813016
Yang W et al (2020a) Clinical characteristics and imaging manifestations of the 2019 novel coronavirus disease (COVID-19):A multi-center study in Wenzhou city, Zhejiang. China Journal of Infection 80:388–393. https://doi.org/10.1016/j.jinf.2020.02.016
Yang X et al (2020b) Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med 8:475–481. https://doi.org/10.1016/s2213-2600(20)30079-5
Yang Y et al (2020c) The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in. China J Autoimmun:102434. https://doi.org/10.1016/j.jaut.2020.102434
Ye M, Wysocki J, William J, Soler MJ, Cokic I, Batlle D (2006) Glomerular localization and expression of angiotensin-converting enzyme 2 and angiotensin-converting enzyme: implications for albuminuria in diabetes. J Am Soc Nephrol 17:3067–3075
Yi Y, Lagniton PNP, Ye S, Li E, Xu R-H (2020) COVID-19: what has been learned and to be learned about the novel coronavirus disease. Int J Biol Sci 16:1753–1766. https://doi.org/10.7150/ijbs.45134
Zanin L, Saraceno G, Panciani PP, Renisi G, Signorini L, Migliorati K, Fontanella MM (2020) SARS-CoV-2 can induce brain and spine demyelinating lesions. Acta Neurochir (Wien) 162:1491–1494. https://doi.org/10.1007/s00701-020-04374-x
Zhang S-Y, Marlier A, Gribouval O, Gilbert T, Heidet L, Antignac C, Gubler MC (2004) In vivo expression of podocyte slit diaphragm-associated proteins in nephrotic patients with NPHS2 mutation. Kidney Intl 66:945–954
Zhang T et al (2016) CaMKII is a RIP3 substrate mediating ischemia-and oxidative stress–induced myocardial necroptosis. Nature medicine 22:175
Zhang C, Shi L, Wang F-S (2020a) Liver injury in COVID-19: management and challenges The lancet. Gastroenterology hepatology 5:428–430
Zhang C, Shi L, Wang F-S (2020b) Liver injury in COVID-19: management and challenges. Lancet Gastroenterol Hepatol 5:428–430. https://doi.org/10.1016/S2468-1253(20)30057-1
Zhang C, Wu Z, Li J-W, Zhao H, Wang G-Q (2020c) The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality International journal of antimicrobial agents:105954
Zhang H et al (2020d) The digestive system is a potential route of 2019-nCov infection: a bioinformatics analysis based on single-cell transcriptomes BioRxiv
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS (2020e) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target Intensive care medicine:1–5
Zheng Y-Y, Ma Y-T, Zhang J-Y, Xie X (2020) COVID-19 and the cardiovascular system Nature Reviews Cardiology:1–2
Zou X, Chen K, Zou J, Han P, Hao J, Han Z (2020) Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med 1–8
Funding
No funding to declare.
Author information
Authors and Affiliations
Contributions
TM and GH conceived of the presented idea. TM, FH, NG, BE, and AY equally participated in drafting the article. TM, and GH participated in revising it critically for essential intellectual content. All authors read and gave approval of this version to be submitted and any revised version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declared no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mokhtari, T., Hassani, F., Ghaffari, N. et al. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J Mol Hist 51, 613–628 (2020). https://doi.org/10.1007/s10735-020-09915-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10735-020-09915-3