Article Text

Abstract
Background: Degenerative lumbar spinal stenosis (LSS) is usually caused by disc herniation or degeneration. Several genetic factors have been implicated in disc disease. Tryptophan alleles in COL9A2 and COL9A3 have been shown to be associated with lumbar disc disease in the Finnish population, and polymorphisms in the vitamin D receptor gene (VDR) (FokI and TaqI), the matrix metalloproteinase-3 gene (MMP-3) and an aggrecan gene (AGC1) VNTR have been reported to be associated with disc degeneration. In addition, an IVS6-4 a>t polymorphism in COL11A2 has been found in connection with stenosis caused by ossification of the posterior longitudinal ligament in the Japanese population.
Objective: To study the role of genetic factors in LSS.
Methods: 29 Finnish probands were analysed for mutations in the genes coding for intervertebral disc matrix proteins, COL1A1, COL1A2, COL2A1, COL9A1, COL9A2, COL9A3, COL11A1, COL11A2, and AGC1. VDR and MMP-3 polymorphisms were also analysed. Sequence variations were tested in 56 Finnish controls.
Results: Several disease associated alleles were identified. A splice site mutation in COL9A2 leading to a premature translation termination codon and the generation of a truncated protein was identified in one proband, another had the Trp2 allele, and four others the Trp3 allele. The frequency of the COL11A2 IVS6−4 t allele was 93.1% in the probands and 72.3% in controls (p = 0.0016). The differences in genotype frequencies for this site were less significant (p = 0.0043).
Conclusions: Genetic factors have an important role in the pathogenesis of LSS.
- single nucleotide polymorphism
- collagen
- intervertebral disc
- stenosis
- AGC1, gene coding for aggrecan
- COL1A1 and COL1A2, genes coding for collagen I α1 and α2 chains
- COL2A1, gene coding for collagen II α1 chain
- COL9A1, COL9A2, and COL9A3, genes coding for collagen IX α1, α2, and α3 chains
- COL11A1 and COL11A2, genes coding for collagen XI α1 and α2 chains
- IVS, non-coding intronic sequence located between coding sequences
- CS, chondroitin sulphate
- CSGE, conformation sensitive gel electrophoresis
- CT, computed tomography
- LDD, lumbar disc disease
- LSS, lumbar spinal stenosis
- MMP, matrix metalloproteinase
- MRI, magnetic resonance imaging
- OLF, ossification of the ligamentum flavum
- OPLL, ossification of the posterior longitudinal ligament
- PCR, polymerase chain reaction
- RT, reverse transcriptase
- SNP, single nucleotide polymorphism
- VDR, vitamin D receptor gene
- VNTR, variable number of tandem repeats
Statistics from Altmetric.com
- AGC1, gene coding for aggrecan
- COL1A1 and COL1A2, genes coding for collagen I α1 and α2 chains
- COL2A1, gene coding for collagen II α1 chain
- COL9A1, COL9A2, and COL9A3, genes coding for collagen IX α1, α2, and α3 chains
- COL11A1 and COL11A2, genes coding for collagen XI α1 and α2 chains
- IVS, non-coding intronic sequence located between coding sequences
- CS, chondroitin sulphate
- CSGE, conformation sensitive gel electrophoresis
- CT, computed tomography
- LDD, lumbar disc disease
- LSS, lumbar spinal stenosis
- MMP, matrix metalloproteinase
- MRI, magnetic resonance imaging
- OLF, ossification of the ligamentum flavum
- OPLL, ossification of the posterior longitudinal ligament
- PCR, polymerase chain reaction
- RT, reverse transcriptase
- SNP, single nucleotide polymorphism
- VDR, vitamin D receptor gene
- VNTR, variable number of tandem repeats
Spinal stenosis (MIM 152550) is defined as a narrowing of the spinal canal, nerve root canals, or intervertebral foramina. It can be congenital or acquired.1 Congenital stenosis is due to an idiopathic developmental narrowing of the spinal canal, typically associated with achondroplasia (MIM 100800) and hypochondroplasia (MIM 146000),2–4 while acquired or degenerative stenosis is usually caused by disc herniation or degenerative changes in the intervertebral discs, facet joints, and ligamentum flavum, but may also be caused by spondylolisthesis, tumours, Paget’s disease or ossification of the posterior longitudinal ligament (OPLL) of the spine.1,3,5,6 Although congenital stenosis is rare, degenerative stenosis is one of the most common clinically important spinal disorders in the aging population.3
Lumbar spinal stenosis (LSS) typically affects subjects over 50 years of age, with a prevalence of 1.7–10%.7,8 Symptoms can be unilateral or bilateral and can include pseudoclaudication, low back pain, numbness, weakness, and pain on extension of the lumbar spine,3,9–11 and can generally be relieved by flexing of the lumbosacral spine.3,9 The correlation of radiographic abnormalities with clinical findings and signs is extremely important for establishing the diagnosis.3,10
Because congenital stenosis is typically associated with chondrodysplasias, which are genetic disorders, it is possible that genetic factors may also play a part in degenerative stenosis. This hypothesis is supported by various findings. Varughese and Quartey reported a case of four brothers with lumbar disc herniation associated with narrowing of the lumbar spinal canal.12 OPLL is a leading cause of spinal stenosis in the Japanese, with a prevalence of 1.9–4.3%.5 A linkage was identified between OPLL and chromosome 6p,6 a region containing the gene for the α2 chain of collagen XI, COL11A2, and an allelic association has been reported between OPLL and a COL11A2 polymorphism, IVS6−4 a>t.6,13 In addition, tryptophan alleles in COL9A2 (Trp2)14 and COL9A3 (Trp3)15 have been shown to be associated with lumbar disc disease (LDD) in the Finnish population. Likewise, a heterozygous mutation resulting in haploinsufficiency of the aggrecan gene, AGC1 was found to lead to intervertebral disc herniation and degeneration in mice,16 and an association between a VNTR in AGC1 and lumbar disc degeneration was subsequently reported in humans.17 Polymorphisms in the vitamin D receptor gene, VDR,18–20 and the matrix metalloproteinase-3 gene, MMP-3,21 have also been shown to be associated with intervertebral disc degeneration.
In view of the evidence for the contribution of genetic factors to lumbar disc disease, disc degeneration and OPLL, we analysed 29 probands with degenerative LSS for sequence variations in nine candidate genes coding for intervertebral disc matrix proteins: COL1A1, COL1A2, COL2A1, COL9A1, COL9A2, COL9A3, COL11A1, COL11A2, and AGC1. In addition, two single nucleotide polymorphisms (SNP) in VDR and one in MMP-3 were analysed.
MATERIALS AND METHODS
Subjects
Twenty nine consecutive Finnish probands (14 male, 15 female) aged from 42 to 75 years (mean 59) with symptoms consistent with LSS were included in the study. The probands were from the Department of Physical Medicine and Rehabilitation, Oulu University Hospital (Oulu, Finland). The symptoms consisted of self reported pseudoclaudication, stenotic symptoms extending to the lower extremities upon extension of the lumbar spine, or numbness or weakness of the lower extremities. The probands were examined clinically and radiologically to confirm the degenerative nature of the stenosis.
Neurological examination included an evaluation of motor reflexes in the lower extremities. Peripheral pulses were evaluated to exclude vascular claudication. The magnetic resonance imaging (MRI) examinations, performed with a 1.5 T scanner (Signa, GE Medical systems), consisted of T2 weighted sagittal fast spin echo images with a repetition time/echo time of 4000/95 ms and a slice thickness of 4 mm. T1 weighted (580/15 ms) sagittal spin echo images and transaxial T2 weighted (6000/105 ms) fast spin echo images were taken, also with a slice thickness of 4 mm. The transaxial MRI images consisted of scans through the L2 to S1 interspaces, and the CT images (Hi Speed Advantage, GE Medical Systems) of scans through the same interspaces with a slice thickness of 4 mm. MRI scans were obtained for all the probands and computed tomography (CT) scans for 25 of the 29 probands (table 1).
Clinical and radiological findings of the probands
The degrees of stenosis, degeneration, and end plate changes were evaluated from the MRI scans. Stenosis was graded as 0 (no stenosis), grade 1 (mild to moderate) if the sagittal diameter of the dural sac at the disc level was from 5 to 10 mm, and grade 2 (severe) if the diameter was <5 mm. Degeneration was evaluated from the T2 weighted sagittal scans and classified as 0 (no signal changes), 1 (slight decrease in signal intensity in the nucleus), 2 (hypointense nucleus pulposus with normal disc height), and 3 (hypointense nucleus with disc space narrowing). End plate changes were graded according to the criteria of Modic et al.22 Ossification of the ligamentum flavum (OLF) was evaluated from the CT scans, on the basis of hyperdense calcified areas in the ligamentum, and OPLL as local calcification on the CT and MRI scan and a hypointense hypertrophic band at the posterior edge of the vertebral body on the T2 weighted sagittal MRI scan. All the MRI and CT scans were read by two experienced doctors who were unaware of the results of the genetic analysis and of the clinical history and physical status of the probands. Those responsible for the clinical assessments were also unaware of the results of the genetic analysis.
The control set consisted of 56 subjects aged 22–72 years (mean 43) with no history of back problems. The control set for the AGC1 VNTR comprised 153 subjects aged 40–45 years with no history of back problems. All controls were Finnish and they were from the same region as the probands.
The study was approved by the local ethics committees. Signed informed consent was obtained from all the subjects.
DNA analysis
Genomic DNA extracted from the blood samples was used as a template for the polymerase chain reaction (PCR). Exons and exon boundaries of COL1A1, COL1A2, COL2A1, COL9A1, COL9A2, COL9A3, COL11A1, COL11A2, and AGC1 (table 2) were amplified by PCR and analysed by conformation sensitive gel electrophoresis (CSGE) as previously described.35 The PCR products that contained heteroduplexes were sequenced with an ABI PRISM 3100 sequencer and BigDye Terminator Sequencing Kit (Applied Biosystems) to define the underlying sequence variations.
Genes and polymorphisms analysed
RNA analysis
Total RNA for proband 23 and a control subject was extracted from Epstein-Barr virus-transformed lymphoblasts. The cDNA synthesis was carried out with the Superscript First Strand Synthesis System for reverse transcriptase (RT)-PCR (Invitrogen, Carlsbad, CA, USA) and followed by amplification with two sets of PCR primers. The first PCR was performed with a pair of primers that corresponded to exons 23 and 30 (5′-CAA GGC GAG AGG GGT CCA GTG and 5′-CAC GGC GAC CTC TGC CAG TTG C) of COL9A2, and the second with a pair that corresponded to exons 24 and 29 (5′-GCT TGC CAG GCG TCA AAG GAG and 5′- CAG CAT CTT CAG CGC CAC ATC). The primer pairs for the COL11A2 cDNA analysis corresponded to exons 5 and 9, the pair used for the first amplification being 5′-CAT GTG AAC AGA AGG AGC TGG A and 5′-AGG TTC CAA CAC TGC AGG CTC and the nested pair 5′-CAT CAA GAC TTC ACA GGC CAC AA and 5′-CTT TCT CTC CCT TCA GCC CTC GG. The amplifications were carried out in a volume of 25 µl containing 1 µl of the cDNA, 7.5 pmol of each primer, 1.5 mM MgCl2, 0.2 mM dNTPs, and 1.5 U AmpliTaq gold DNA polymerase. The conditions, after an initial denaturation at 95°C for 12 minutes, were 34 cycles of 30 seconds at 95°C, 30 seconds at 60°C and 30 seconds at 72°C, followed by a final extension at 72°C for 8 minutes. The products were analysed on agarose gels and by sequencing.
Southern analysis of the AGC1 VNTR
A probe for the AGC1 VNTR in exon 12 was generated by PCR.31 The PCR was carried out with the Advantage-GC Genomic PCR kit (Clontech, Palo Alto, CA, USA) with 0.5 M of GC-melt mix. The PCR product was analysed on agarose gel, purified from the gel (Qiaex II Gel Extraction Kit, Qiagen, Valencia, CA, USA), cloned, and sequenced using the ABI PRISM 377 Sequencer (Applied Biosystems). The clone was digested with EcoRI, and labelled with [32P]dCTP using the Rediprime II DNA labelling system (Amersham Pharmacia Biotech, Piscataway, NJ, USA).
VNTR polymorphism was analysed by Southern hybridisation.31 Five µg of genomic DNA was digested with HaeIII and separated on a 1.2% agarose gel for 20 hours at 45 V. A molecular weight marker, a 100 bp DNA step ladder (Promega, Madison, WI, USA), was added to each gel. The size of one repeat was 57 bp. The number of repeats for each sample was estimated from the sizes of the fragments observed in Southern analysis.
Analysis of VDR and MMP-3 polymorphisms
Two intragenic polymorphisms, T to C in exon 918 and T to C at the translation initiation codon36,37 in VDR were analysed as previously described. Exon 9 T to C polymorphism was detected by TaqI restriction enzyme digestion. The alleles were named T and t, the capital denoting absence of the TaqI site or nucleotide T. The T to C polymorphism at the translation initiation codon (ATG/ACG) of VDR was determined using the FokI restriction enzyme. Allele F indicates the presence of nucleotide C, which results in the absence of the first translation initiation codon, and use of the second methionine codon located three amino acids downstream.36,37
A 179 or 180 bp region of the MMP-3 promoter containing a 5a/6a polymorphism was determined by sequencing (table 2).21
Statistical analysis
To test the equality of the allele frequencies between the probands with LSS and controls, χ2 tests were carried out for 2×2 tables of allele counts. Haplotype frequencies for four marker loci, COL11A2 IVS6−4, VDR (FokI and TaqI), and MMP-3 polymorphisms, were estimated with the snphap program, which applies maximum likelihood estimation via a so-called EM algorithm.38 A likelihood ratio test was employed to test for differences in these frequencies between the probands and controls.
RESULTS
Clinical and radiological findings
All 29 probands with degenerative LSS who were examined clinically and radiologically had a self reported limitation in walking distance (range 50–3000 m, table 1) and at least one other stenotic symptom (pain or numbness of the lower extremities upon extension of the lumbar spine, or numbness or weakness in the lower extremities). The clinical findings showed seven probands with bilateral tendon reflex defects in the lower extremities, but none had objective muscle weakness or peripheral pulse abnormalities. Twelve probands had undergone a decompressive operation. In addition, three (Nos 2, 20, and 24) had previously been operated on for a herniated nucleus pulposus and were found to have a stenosis at a different lumbar level (table 1).
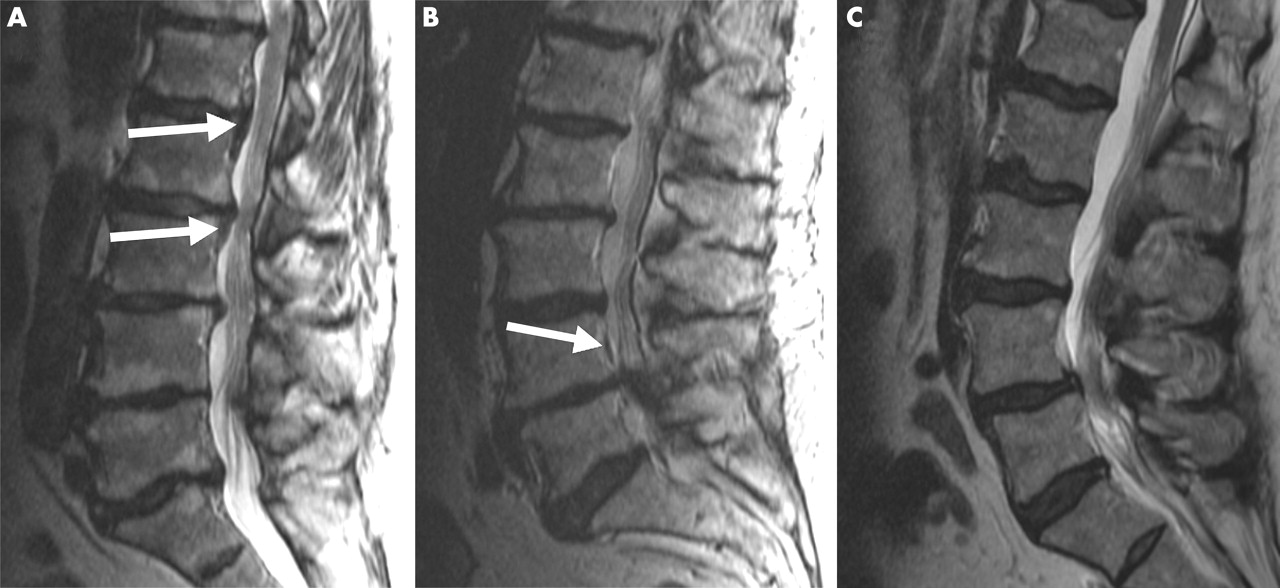
Radiological evaluation of the lumbar region indicated that every proband, except for No 13, who had undergone decompressive surgery for stenosis before inclusion in the study, had stenosis at least at one level (table 1). Severe disc degeneration (grade 2 or 3) was also seen in every proband at least at one lumbar level (table 1, figs 1A, B, and C). Twenty probands had grade 2 end plate degeneration according to Modic’s criteria (table 1, figs 1A and 1B), six had OPLL (table 1, fig 1B), and eight were found to have OLF (table 1).

T2 weighted transaxial MRI scans (TR 6000 ms/TE 105 ms) of probands 2 (A), 10 (B), and 23 (C). (A) A hypertrophic band is seen at the L2–3 disc level and caudally at L3–4 (white arrows). Both disc and end plate degeneration are seen at all lumbar levels. (B) The scan shows OPLL at the L4 vertebral level (white arrow). This proband also had severe disc and end plate degeneration at multiple lumbar levels. (C) The scan indicates severe disc degeneration at multiple lumbar levels, and a Schmorl’s node at L2–3.
DNA analysis of collagen genes and AGC1
The probands were analysed for mutations in COL1A1, COL1A2, COL2A1, COL9A1, COL9A2, COL9A3, COL11A1, COL11A2, and AGC1 (table 2).
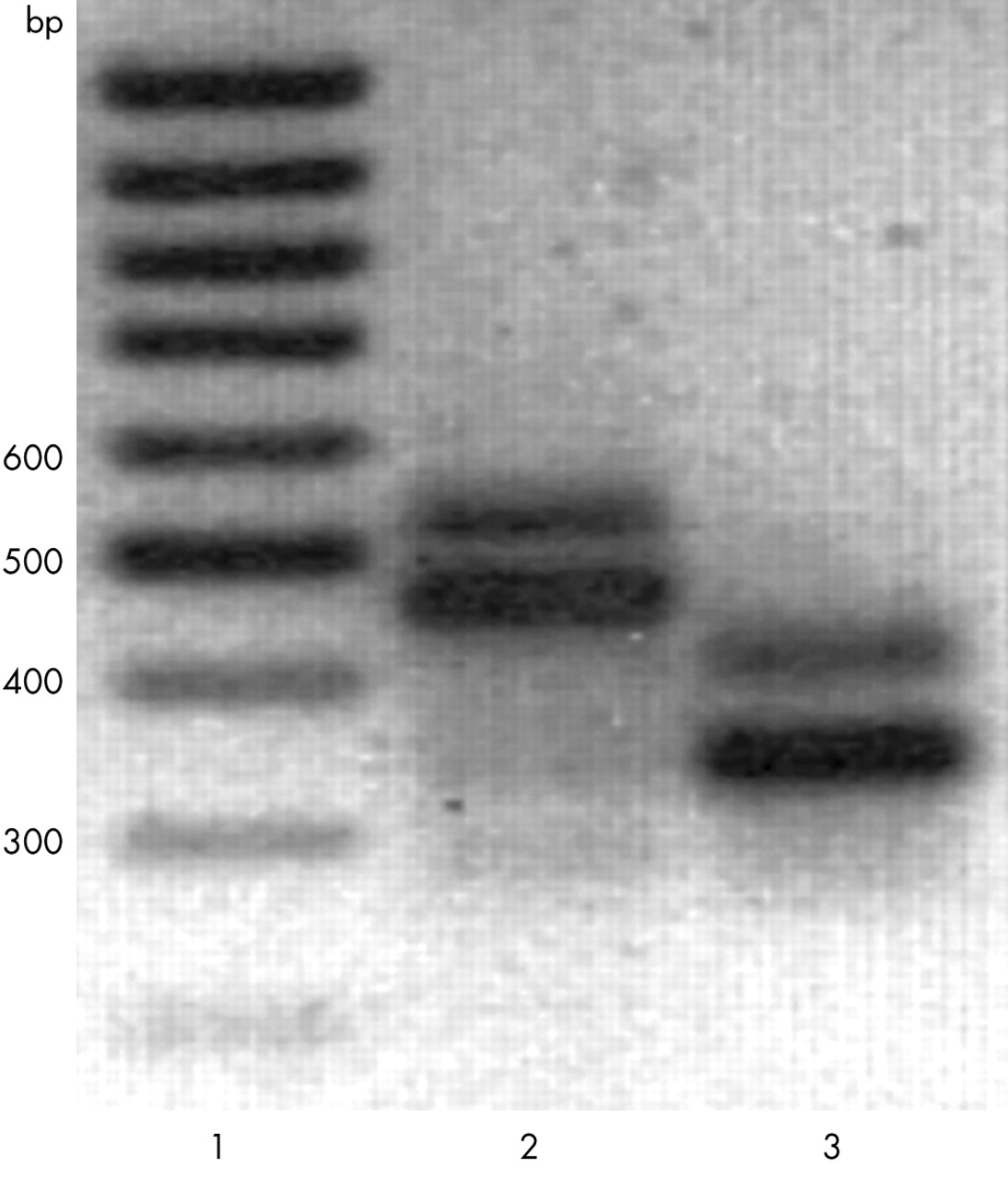
Analysis of the collagen IX genes, COL9A1, COL9A2 and COL9A3, showed three interesting changes (table 3): an a to c change in COL9A2 IVS26−2, Gln326 to Trp (Trp2 allele) in the α2(IX) chain,14 and Arg103 to Trp (Trp3 allele) in the α3(IX) chain.15 Proband 23 (table 1) had the a to c mutation in COL9A2 IVS26−2. This mutation was not found in the controls. Total RNA was isolated from the lymphoblasts of proband 23 and analysed by RT-PCR for possible splicing defects, resulting in the identification of two products, of about 550 bp and 460 bp with the primer set for exons 23–30, and of about 430 and 340 bp with the primer set for exons 24–29 (fig 2). Sequencing indicated that the smaller product contained only the wild-type sequence, while the larger product contained the wild-type sequence and sequences for the entire intron 26 (not shown). The mutation prevented splicing of intron 26, leading to an insertion of 88 extra nucleotides, and thus of 21 new amino acids, followed by a premature stop codon. The proband’s 33 year old son and 87 year old mother were clinically unaffected and did not carry the mutation. The proband’s father was not available for examination.
Genetic findings of the probands
{kind=link}
{kind=link}
Agarose gel electrophoresis of α2(IX) RT-PCR products. Total RNA from proband 23 was analysed by RT-PCR using primer pairs corresponding to exons 23 and 30 (lane 2), or exons 24 and 29 (lane 3), as indicated in “Materials and methods”. The analysis showed two products of about 550 bp and 460 bp (lane 2) and 420 bp and 340 bp (lane 3). Sequencing indicated that the lower molecular weight products contained the wild-type sequence, whereas the higher molecular weight products also contained intron 26. Lane 1 contains a 100 bp ladder.
Proband 20 was heterozygous for the Trp2 allele. He had been previously operated on for a herniated nucleus pulposus, but also underwent a decompressive operation at a different level during the course of the present study. Probands 14, 26, and 27 were heterozygous for the Trp3 allele and proband 1 was homozygous for it.
An a to t polymorphism in IVS6−4 of COL11A2 has been reported to be associated with OPLL in the Japanese population.13 Even though OPLL is not a common cause of spinal stenosis in the Finnish population and was not a common finding among the probands (table 1), the possibility of an association between this polymorphism and stenosis was studied. The analysis showed that the frequency of the t allele was 93.1% in the probands but only 72.3% in the control group (p = 0.0016; tables 3 and 4), although the genotype frequency differences were less significant (p = 0.0043). Based on this significant result (table 4), we wanted to see whether one of the genotypes exerts a dominant effect. Indeed, the t/a and a/a genotypes did not occur in significantly different proportions in the proband and control subjects while the t/t genotype was found much more frequently in the probands than in the controls. Thus, we combined the t/a and a/a genotypes (4 probands, 28 controls) and contrasted it with the high risk t/t genotype (25 probands, 28 controls, p = 0.0011). The significance of our finding of the high risk t/t genotype (the “a” allele may also be viewed as protecting) may be expressed as the population attributable risk 39, λ = 1.98—that is, the disease in an estimated 20% of the affected subjects in the population is attributable to the tt genotype.
Allele and genotype frequencies
Another polymorphism, exon 6+28A>G, also reported previously,13 was found to be in complete linkage disequilibrium with the IVS6−4a>t polymorphism. The same authors have shown that the IVS6−4 a allele is associated with skipping of exon 6 and retention of exon 7. We confirmed this finding here by performing an RT-PCR analysis on total lymphoblast RNA from proband 23, who was homozygous for the IVS6−4 t allele. The analysis showed only one fragment, and contained sequences for exon 6 (78 bp) and 7 (63 bp), but exon 8 (180 bp) was not present (not shown).
A number of sequence variations were detected in all collagen genes and AGC1, but they were likely to be neutral, because they were found in equal frequencies in the controls (not shown).
Southern analysis of the AGC1 VNTR
An association between an AGC1 VNTR polymorphism and LDD has been reported previously, in a study that suggested an overrepresentation of alleles with small numbers of the repeats in subjects with multilevel disc degeneration.17 Because disc degeneration is a common cause of stenosis and was present in all the probands, the VNTR polymorphism was analysed here (table 2). The alleles with 27 and 28 repeats were found to be the most common in both the stenosis (table 3) and the control group (not shown), and there were no significant differences in allele frequencies between the two groups.
VDR and MMP-3 polymorphisms
Two VDR polymorphisms reported to be associated with disc degeneration were studied: FokI and TaqI. FokI restriction enzyme digestion of exon 2 of VDR showed that the frequency of the f allele was 41% in the probands and 32% in the controls (tables 3 and 4), a difference that was not significant (p = 0.24), while the allele frequencies for TaqI polymorphism likewise did not differ significantly between the proband and control groups (tables 3 and 4).
The 5a/5a and 5a/6a genotypes of the MMP-3 promoter region have been reported to be associated with intervertebral disc degeneration.21 No significant differences were found in the allelic frequencies or genotype distributions between the probands and controls (tables 3 and 4).
Haplotype analysis
Of the 16 possible haplotypes at the four sites, 13 (almost all) were observed in these data, which indicated a weak association between the sites (linkage disequilibrium). The haplotype frequencies did not differ significantly between the probands and controls (p = 0.076).
DISCUSSION
LSS is common and one of the most clinically important spinal disorders.7,8 Because a number of genetic factors have been implicated in disc herniation, disc degeneration, and OPLL, which are the leading causes of degenerative stenosis,6,13,40 we investigated possible associations between the previously identified genetic factors and degenerative LSS, and also analysed nine genes encoding intervertebral disc matrix proteins for sequence variations.
An association has been reported between OPLL and the COL11A2 IVS6−4 t allele in the Japanese population.6,13,41 The present screening of all exons and boundaries of COL11A2 for mutations in probands with LSS did not identify any unique sequence variations or putative causative mutations, but the frequency of the IVS6−4 t allele was found to be 93.1% in the probands compared with 72.3% in the controls (p = 0.0016). The corresponding allele frequencies for OPLL positive and negative subjects reported by Koga et al were 86% and 74%.6 Thus our results further strengthen the hypothesis of a role for COL11A2 in stenosis, and also suggest that the IVS6−4 t allele can predispose carriers to degenerative LSS, which is not commonly associated with OPLL. It is not clear, however, by what mechanism this predisposition operates. COL11A2 exons 6 to 8 undergo complex alternative splicing, and Maeda et al have shown that the IVS6−4 a allele is associated with a different splice pattern from the t allele, exon 6 being skipped in its presence.13 Our present observations are in agreement with this finding. In addition to the inclusion of exon 6, we showed here that exon 8 was skipped in the presence of the t allele. Because any nucleotide can be found at position −4 in the acceptor splice site,42 the causal relationship between the alternatively spliced forms and the two alleles is not obvious, and, possibly, the factor responsible for the observed splice variants is either exon 6+28A>G polymorphism, which is in complete linkage disequilibrium with the IVS6−4a>t polymorphism, or another, yet unidentified variation.
A role for collagen IX in spinal disorders is supported by human14,15 and animal studies.43,44 Further evidence is presented in our study. One of the present probands, a subject with a history of LDD, had the Trp2 allele and four had the Trp3 allele. Because LDD is a common cause of LSS, it is possible that these alleles also predispose its carriers to stenosis. In the light of our previous findings, it is not likely that the Trp3 allele alone will lead to LSS, but it may be one of the predisposing factors. In addition, the analysis identified a heterozygous a to c mutation in IVS26−2 of COL9A2. The mutation prevented splicing of intron 26 and resulted in an insertion of 21 new amino acids followed by a premature translation termination that was derived from intron 26. It is predicted that this mutation will result in synthesis of a truncated α2(IX) chain that lacks the C-terminal end required for the assembly of the collagen IX heterotrimer of three α chains; the mutation is likely to lead to a reduced amount of collagen IX in the tissue. This is the first report of a splicing mutation in collagen IX leading to a premature translation termination and affecting the C-terminal end of the molecule.
Several predisposing genetic factors have been identified in disc degeneration. One of them is the VNTR polymorphism in AGC1. This is located in the chondroitin sulphate binding region (CS1), which is composed of repeated sequences of 57 nucleotides or 19 amino acids, the number of repeats varying from 13 to 33.31 It has recently been shown that subjects with a small number of repeats have an increased risk of disc degeneration, presumably because aggrecan molecules with fewer CS chains will have a poorer ability to hydrate the disc.17 No association between the VNTR polymorphism and LSS was found here, however, nor was any found between LSS and the VDR and MMP-3 polymorphisms, which have also been implicated in disc degeneration.18–21 These findings suggest that disc degeneration is highly heterogeneous both clinically and genetically, and that leading to spinal stenosis may represent a distinct subgroup.
The genes coding for collagens I and II, which are the major intervertebral disc components, were also analysed. Mutations in these genes are known to cause a variety of osteochondrodysplasias,45 but their role in spinal diseases has not been investigated previously. The analysis did not provide any evidence that sequence variations in these genes are associated with spinal stenosis.
Despite the small sample size this study provides evidence that genetic factors are important in the causation of LSS. However, a larger population sample is needed to confirm these preliminary findings.
Acknowledgments
We thank Ms Aira Harju (Collagen Research Unit, Biocentre and Department of Medical Biochemistry and Molecular Biology, Oulu, Finland), Mr Hannu Hietala, Ms Christina L Troxell, and Ms Jaana Väisänen (Center for Gene Therapy and Department of Medicine, Tulane University Health Sciences Center, New Orleans, Louisiana, USA) for their expert technical assistance.
This work was supported by grants from the National Institute of Health (AR45982 to L A-K, HG00008 to JO), the Academy of Finland, the Louisiana Gene Therapy Research Consortium (New Orleans, LA) and HCA-The Health Care Company (Nashville, TN) (to LA-K).