Article Text

Abstract
Objective: To examine the influence of genotype on late gadolinium enhancement (LGE) and the potential of cardiovascular magnetic resonance (CMR) to detect preclinical hypertrophic cardiomyopathy.
Design: Prospective, blinded cohort study of myocardial LGE in a genetically homogeneous population.
Patients: 30 patients with disease causing mutations in the recognised hypertrophic cardiomyopathy gene for cardiac troponin I (TNNI3): 15 with echocardiographically determined left ventricular hypertrophy (LVH+) and 15 without (LVH−).
Main outcome measures: CMR measures of regional left ventricular function, wall thickness, and mass, and the extent and distribution of LGE.
Results: LGE was found in 12 (80%) LVH+ patients but with variable extent (mean 15%, range 3–48%). LGE was also found in two (13%) LVH− patients but the extent was limited (3.6%) and both patients were found to have an abnormal ECG and regional hypertrophy by cine CMR. The extent of LGE was positively associated with clinical markers of sudden death risk (21% with ⩾ 2 risk factors v 7% with ⩽ 1 risk factor, p = 0.02) and left ventricular mass (r = 0.56, p < 0.001) and was inversely associated with ejection fraction (r = −0.58, p < 0.001). Segmental analysis showed that as regional wall thickness increased, LGE was more prevalent (p < 0.0001) and more extensive (r = 0.98, p = 0.001).
Conclusion: In patients with disease causing mutations in TNNI3, focal fibrosis was not detected by LGE CMR before LVH and ECG abnormalities were present. Once LVH is present, LGE is common and the extent correlates with adverse clinical parameters. This suggests that focal fibrosis is closely linked to disease development.
- CMR, cardiovascular magnetic resonance
- DTPA, diethylenetriaminepentaacetic acid
- HCM, hypertrophic cardiomyopathy
- LGE, late gadolinium enhancement
- LVH, left ventricular hypertrophy
- hypertrophic cardiomyopathy
- magnetic resonance imaging
- fibrosis
- gadolinium
Statistics from Altmetric.com
- CMR, cardiovascular magnetic resonance
- DTPA, diethylenetriaminepentaacetic acid
- HCM, hypertrophic cardiomyopathy
- LGE, late gadolinium enhancement
- LVH, left ventricular hypertrophy
Hypertrophic cardiomyopathy (HCM) is a clinically and genetically heterogeneous disease. HCM can be defined in a variety of ways: genetically by the presence of disease causing mutations in any of 10 genes encoding sarcomeric proteins1; and clinically by the presence of unexplained cardiac hypertrophy.2 Histologically, HCM is characterised by the presence of extensive myocardial disarray and various patterns of myocardial fibrosis.3,4 The link between genetic mutation and phenotype development including risk of sudden death and the development of heart failure is poorly understood but the environment, sex, and modifier genes5,6 are important in causing progressive myocardial abnormality characterised by hypertrophy, myocardial fibrosis, and disarray.7,8
It has not been possible to comprehensively quantify myocardial fibrosis and disarray in vivo9 but a new technique, late gadolinium enhancement (LGE) cardiovascular magnetic resonance (CMR), shows promise in this regard.10,11 Regions of myocardial LGE, found after a gadolinium contrast agent injection, represent regions of increased myocardial fibrosis, and allow a direct assessment of the underlying myocardial abnormality.12 Previous studies in patients with ungenotyped HCM have shown correlations of LGE extent with clinical risk of sudden death and presence of heart failure.10 However, it is not known whether in these studies the LGE found reflects primarily genetic or non-genetic contributions or whether LGE would be useful for the detection of early, preclinical disease.
In this study, we examined a genotyped cohort, all of whom carried mutations in the cardiac troponin I gene (TNNI3).13 We hypothesised that the presence of LGE would be useful to detect early disease and would remain linked to clinical risk of sudden death despite the presence of mutations in the same disease causing gene.
METHODS
Genotyped patients
As part of an ongoing gene identification programme in an established HCM clinic, 748 unrelated patients were screened for mutations in the cardiac troponin I gene, as previously reported.13 A total of 23 families were identified with 100 mutation carriers, 48 of whom fulfilled established diagnostic criteria for HCM.2 The evidence that the mutations are disease causing was previously published and is essentially as follows: firstly, the amino acid substitution identified in probands was present in all relatives with the disease; secondly, no sequence variations led to amino acid substitutions in 150 ethnically matched control chromosomes; thirdly, identical mutations appeared in several unrelated families; and lastly, all mutations were located in functionally important and conserved regions of the gene.12–15 All living patients without contraindications to CMR in the UK from this cohort were invited to participate. Patients with all 13 mutations within the main study participated. Thirty patients were recruited: 15 with unexplained left ventricular hypertrophy (LVH) on echocardiography (LVH+) and 15 gene positive relatives without (LVH−).
Patient phenotype characterisation
All patients at entry into the HCM clinic underwent 12 lead ECG, two dimensional echocardiography, and stratification for risk factors for sudden death according to five standard criteria. Five clinical risk factors for sudden death were used to stratify patients: a family history of HCM and two or more family members with sudden premature (⩽ 40 years) cardiac death; unexplained syncope; non-sustained ventricular tachycardia defined as three or more ventricular extrasystoles at a rate of 120 beats/min on ambulatory ECG; an abnormal blood pressure response (defined as a failure of systolic blood pressure to rise by more than 25 mm Hg from baseline values or a fall of more than 15 mm Hg from the maximum blood pressure) during upright exercise testing in ⩽ 40 year olds; and the presence on echocardiography of severe LVH (⩾ 30 mm).16 This assessment was repeated at entry into this study to assess disease progression and to ensure correct stratification into LVH+ or LVH− categories. All echocardiography was performed by experienced operators in the dedicated HCM clinic and reviewed at entry into the study.
CMR
CMR was performed as previously described.10 Briefly, a short axis stack of cine images was acquired to assess global and regional function. A bolus of 0.1 mmol/kg gadolinium-diethylenetriaminepentaacetic acid (DTPA) was administered intravenously and LGE imaging was started at five minutes with an inversion recovery technique, meticulous adjustment of the inversion time, a 90° pre-saturation pulse over the cerebrospinal fluid to eliminate ghosting, and imaging parameters optimised according to the patient’s breath hold ability and heart rate. Images with potential LGE were repeated with a phase swap for confirmation.
Image analysis
All data were analysed blinded to the clinical details. The cine images were quantified by planimetry to determine myocardial mass. The maximum diastolic segmental wall thickness was measured. When present, LGE was quantified for extent and this was expressed as a percentage of the total myocardial mass. Segments were analysed in a 16 segment (six base, six mid, and four apex) model,17 with both diastolic wall thickness and LGE expressed as quintiles: ⩽ 9 mm; 10–14 mm, 15–19 mm; 20–24 mm; ⩾ 25 mm; no LGE, ⩽ 24%; 25–49%; 50–74%; ⩾ 75%.
Statistical analysis
Non-parametric Wilcoxon rank comparisons were used to compare LGE extent in patients with fewer (⩽ 1) versus more (⩾ 2) risk factors for sudden death. Regression was used to compare total extent of LGE with continuous variables (age, left ventricular mass, ejection fraction, and wall thickness). The segmental presence of LGE and wall thickness were compared by 2 × n contingency table analysis.
RESULTS
Patient baseline characteristics
Table 1 gives patient baseline characteristics (at time of scanning). Eleven patients had asymmetrical hypertrophy (three with outflow obstruction), one in a dilated phase, two with apical HCM, and one with severe restrictive physiology.
Distribution of risk factors for sudden death and clinical characteristics of patients with (LVH+) and without (LVH−) left ventricular hypertrophy
Phenotype development before CMR
During clinic follow up before CMR (mean 49 months), phenotype had changed in five patients. One patient had developed progressive disease with wall thinning and cavity dimension increase. Two patients had developed echocardiographic LVH (in one, maximum left ventricular wall thickness became > 30 mm; both also developed syncope). Two patients who remained LVH− developed clinical risk factors (one syncope and one abnormal exercise blood pressure response).
LGE in LVH+ and LVH− patients
Myocardial LGE was found in 12 (80%) LVH+ patients, with variable extent (mean 15%, range 3–48%). LGE was also found in two (13%) LVH− patients with limited extent (3.6%). Both of these patients and one other LVH− patient were found to have regional hypertrophy by cine CMR despite normal echocardiography. These patients had abnormal Q waves and ST segment abnormalities. All the LVH+ patients also had abnormal ECGs. No other LVH− patient had an abnormal ECG.
LGE and clinical risk
The extent of LGE was associated with clinical risk factors for sudden death (⩾ 2 v ⩽ 1 risk factors, 21% v 7%, p = 0.02) and total left ventricular mass (r = 0.56, p < 0.001) and was inversely associated with ejection fraction (r = −0.58, p < 0.001).
LGE within families
Three families had more than four gene positive members, which illustrate the correlation of LGE, phenotype, and phenotype progression.
Family H805 (Arg145Trp) (fig 1) has late onset, low risk disease. No patient has any risk factors for sudden death. No phenotype development was detected over three years of follow up. Individual 4, the proband, has the most LVH and it is associated with outflow tract obstruction and some (6%) myocardial LGE. Individuals 2 and 3 fulfil proposed familial criteria for HCM with abnormal ECGs but normal echocardiograms. CMR detected LVH in both individual 2 and 3 and limited LGE in individual 3 (3%).
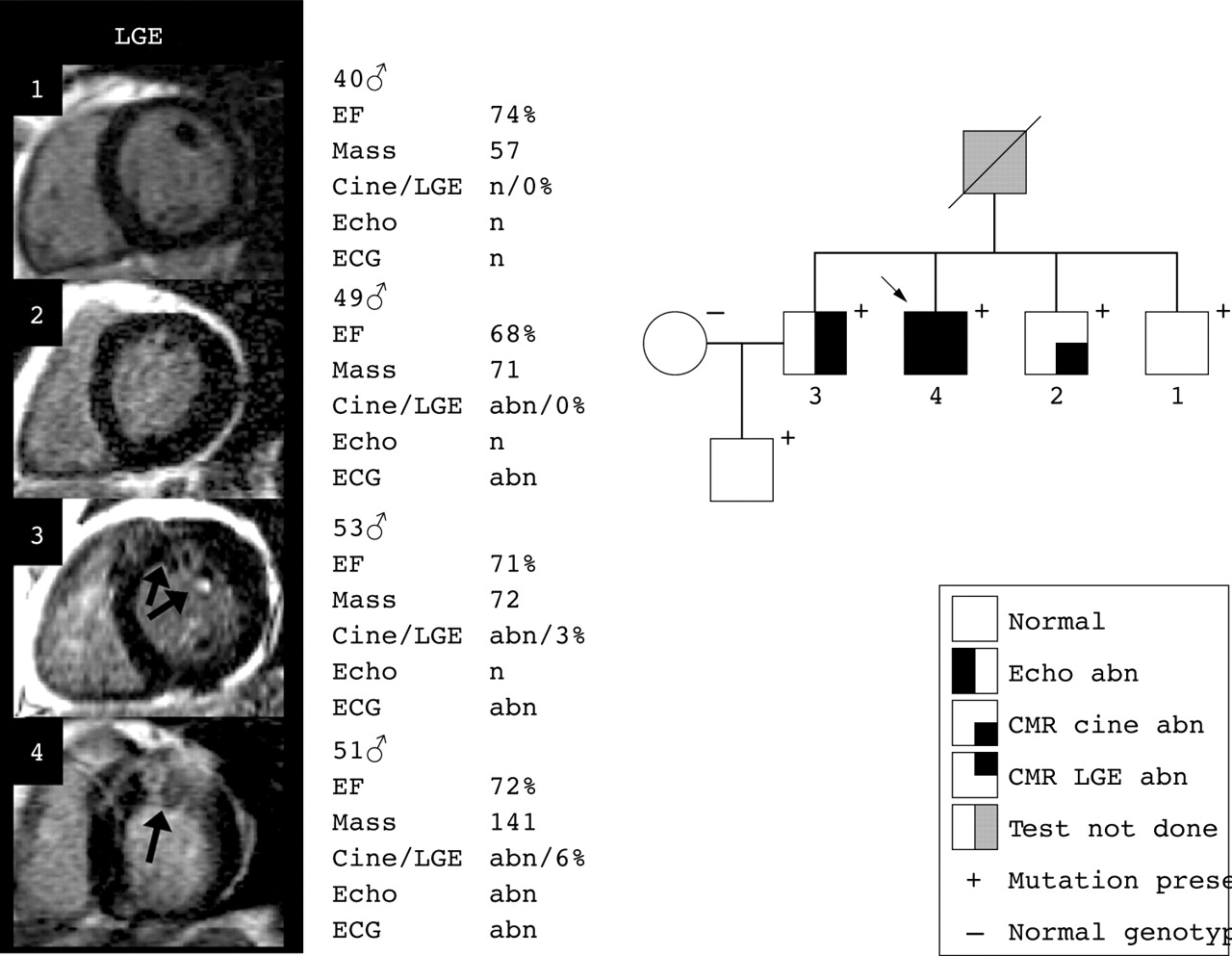
Progression of late gadolinium enhancement (LGE) in a family with more than four gene positive members: H805 (Arg145Trp), a family with late onset, low risk disease without progression during follow up and little LGE. abn, abnormal; CMR, cardiovascular magnetic resonance; EF, ejection fraction; n, normal.
In family H15 (Arg162Gln) (fig 2) the brother (proband) died suddenly aged 18 years and post mortem examination found extensive myocardial fibrosis and asymmetric hypertrophy. Over 12 years’ follow up, individual 4, a male, developed LVH, which progressed to > 30 mm and an abnormal blood pressure response to exercise; 13% LGE was found in the hypertrophied region of the anterior septum. His older sister had phenotype development with an abnormal ECG and an abnormal blood pressure response to exercise. CMR showed mild LVH and 3% LGE.

Progression of LGE in a family with more than four gene positive members. H15 (Arg162Gln), a family with early onset disease (in males) with rapid phenotype development, significant enhancement in the hypertrophied regions, and significant risk of sudden death.
Family H136 (Arg186Gln) (fig 3) has two patients who died in their 40s of heart failure (progressive disease). Individual 3 developed LVH and syncope over six years. CMR showed 11% LGE. Over 14 years’ follow up, individual 4 developed LVH > 30 mm, an abnormal exercise test, and subsequently wall thinning and left ventricular dilatation. CMR showed very extensive LGE of 48%.

Progression of LGE in a family with more than four gene positive members. H136 (Arg186Gln), a family with disease that progressed from normal through hypertrophy to heart failure. There is significant LGE in hypertrophied myocardium and massive LGE in the patient with previous left ventricular hypertrophy > 30 mm and subsequent wall thinning.
Segmental LGE and regional hypertrophy
As regional wall thickness increased, LGE was more prevalent (p < 0.0001 for trend) and more extensive (r = 0.98, p = 0.001) (fig 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Combining the 480 myocardial segments from the 30 patients, the (A) prevalence and (B) extent of LGE increases with increasing segmental wall thickness.
DISCUSSION
Familial genotype–phenotype studies indicate that disease causing sarcomeric gene mutations account for only some of the characteristic phenotypic variation in HCM.18 Studying a cohort with disease causing mutations of a single gene reduces the contribution of the sarcomeric mutation to phenotypic heterogeneity and allows investigation of patients before phenotypically declared disease.
Myocardial fibrosis plays an important part in end stage disease as shown by necropsy studies8,19 but the role of fibrosis in developing disease is not understood because there has been no sensitive in vivo quantification technique. A greater understanding of phenotype development is a major aim in HCM because, although the genetic basis of HCM has been largely elucidated, the mediators between genetic defect and myocardial abnormality, and hence risk, are only now being investigated.6
LGE CMR provides a means of quantifying fibrosis in HCM. Gadolinium-DTPA is an extracellular tracer. After an intravenous bolus it is known that fibrosis from myocardial infarction appears enhanced by the highly sensitive inversion–recovery CMR technique late after injection.20 LGE is also found in HCM11 and has been found to represent regions of interstitial expansion and fibrosis.12,21 It has been shown that the extent of LGE is associated with clinical risk factors for sudden death and the presence of progressive disease.10
The data presented in this paper support the clinical perspective that myocardial fibrosis, detected as LGE, may play an important part in HCM phenotype development. With the use of CMR to define the presence or absence of LVH, regions of LGE were found in 14 of 18 (78%) patients with LVH but in no patient without LVH, suggesting that regions of fibrosis occur only after the development of LVH. This is backed up by the strong but imperfect relation of segmental LGE presence and extent with segmental wall thickness (fig 4). In addition, the total extent of LGE was higher in patients with more risk factors for sudden death—a cohort in whom necropsy studies suggest that advanced myocardial abnormalities can be expected. The within-family data (for example, the family in fig 3), where clinical follow up had documented progressive myocardial hypertrophy and risk factor development, and in one patient subsequent thinning and left ventricular impairment show progressive LGE, even when the wall is thinning. These data suggest that the extent of LGE found in a patient, by being a marker of global myocardial abnormality, may be an indicator of a patient’s risk at the time of measurement, reflecting the influence of all contributing factors, both genetic and environmental. Of course, the relation between extent of fibrosis and events may be complex and we hypothesise that the rate of LGE development may also be important—extensive LGE at a young age may carry more significance than a similar degree of LGE in an older patient.
A further implication of the study is that in patients with a TNNI3 mutation, the detection of LGE by CMR is not useful for screening for early disease before hypertrophy is present. Indeed, the study emphasises the known importance of ECG abnormalities in early disease22,23 because the three patients who were LVH− by echocardiography but LVH+ by CMR had abnormal ECGs. But the finding of a high incidence of LGE (78%) in genotyped HCM is preliminary suggesting that LGE may be a highly specific marker of myopathy, although it will not be 100% sensitive, and may provide a useful technique for distinguishing HCM from other forms of non-myopathic LVH such as compensatory or physiological forms.
A limitation of this study is that we attempted to determine the genetic contribution to HCM but it is clear that there is considerable phenotype heterogeneity with different TNNI3 mutations, so some within-family analysis is presented. There may also be a genetic influence from linked and unlinked genetic modifiers. The number of patients is limited in this study and generalising the results to other gene mutations may need caution. This study brings together a novel technique for myocardial phenotyping, LGE CMR, with a population of patients with HCM who are well characterised both genetically and clinically. Further such studies are needed because LGE CMR has great potential to provide new insights into pathogenesis and disease expression in HCM.
Acknowledgments
Professor McKenna (chair holder), Dr James Moon (Junior Research Fellowship), and the Centre for Advanced Magnetic Resonance in Cardiology (CAMRIC) were supported by the British Heart Foundation. Research support was also received from CORDA and Siemens Medical Solutions.