Article Text

Statistics from Altmetric.com
The sudden death of a parent or sibling is a great tragedy for a child, with many consequences. In the case of proven or suspected sudden cardiac death, one obvious question is whether the same disease is present in the child. There are many causes of sudden cardiac death, only some of which may present a risk to other family members. Identification of pre-symptomatic disease presents the opportunity for a risk assessment and preventative treatment. Confirmation of the absence of the same disease leads to appropriate reassurance.
In the event of sudden death there is a responsibility to offer assessment to other family members—in most cases first degree relatives. It is important to obtain an accurate and complete pedigree to identify individuals likely to benefit from screening. The best approach is to use a stepwise assessment and only to extend screening through the family if other key individuals are identified. It is not appropriate to start with distant relatives—it is common for parents to ask for their children to be screened when they have not been assessed themselves.
The assessment of the family depends fundamentally on the diagnosis of the cause of sudden death. An autopsy will usually provide confirmation of familial diseases such as hypertrophic cardiomyopathy, dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, and aortic rupture in Marfan syndrome, and will also identify non-familial abnormalities such as coronary artery malformations. In the past routine coroners’ autopsies were probably less reliable. The ideal is for the examination to be performed by a specialist cardiac pathologist with appropriate samples being taken for histological and genetic analysis. The autopsy will be unhelpful in primary electrical diagnoses such as long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), Brugada syndrome, etc.1
HYPERTROPHIC CARDIOMYOPATHY
Hypertrophic cardiomyopathy is an important cause of sudden death in the young. It is a familial disease characterised by unexplained left ventricular hypertrophy. The disease is caused by mutations in genes encoding protein components of the cardiac sarcomere. Mutations in any one of at least 10 genes may be responsible and more than 200 individual mutations have been reported—most are believed to be private to individual families.
Although this is a genetic disease, diagnosis usually depends on clinical, ECG, and echocardiographic findings. Genetic testing remains complex, time consuming and is currently not widely available for clinical use. About 60% of patients with the hypertrophic cardiomyopathy phenotype have identifiable mutations.
In the event of a sudden death from hypertrophic cardiomyopathy, first degree relatives should be offered clinical assessment, an ECG, and an echocardiogram. Clinical recognition of gene carriers is complicated by variable penetrance and age-related penetrance. Although hypertrophic cardiomyopathy may be recognised in early childhood, most affected individuals probably have normal echocardiograms early in life and develop left ventricular hypertrophy in late childhood or adolescence. It used to be recommended that a normal echocardiogram at physical maturity predicted a normal genotype but more recently it has been recognised that the phenotype may develop later in adult life.2 It is also well recognised that some gene carriers may not have abnormal ECGs or echocardiograms.
Current recommendations are that individuals with a 1:2 risk of being affected by hypertrophic cardiomyopathy should have echocardiographic screening every 12–18 months from the age of 12 until late teenage and perhaps every five years thereafter.2 However, one should also consider the negative side of recommending regular screening of adults who are very likely to be normal, such as effects on employment, insurance, etc. If the diagnosis is first recognised after sudden death it is probably preferable also to include younger children in the initial screening and then to enrol them in regular screening from the age of 12.
Assessment of the ECG and echocardiogram in childhood can be difficult unless the findings are very obviously abnormal.3 At any age it is very difficult to define an “upper limit of normal” for echocardiographic measurements of left ventricular wall thickness and minor, non-specific variations in the ECG are relatively common. Confirmation of hypertrophic cardiomyopathy should prompt more formal risk assessment. Diagnosis of hypertrophic cardiomyopathy during childhood as a result of family screening is relatively uncommon. For example, only 23% of children with hypertrophic cardiomyopathy in a report from Toronto were identified through family screening.4 In the same report a family history of sudden death was not predictive of a higher risk in children identified through screening.
Family history of sudden death from hypertrophic cardiomyopathy
-
Clinical examination, ECG and echocardiogram in all first degree relatives.
-
Repeat echo/ECG
at age 12 years
every 12–18 months until maturity
possibly every five years thereafter
-
Refer children with abnormal ECG or echocardiogram for risk assessment
-
Consider genotyping the family if available
DILATED CARDIOMYOPATHY
Dilated cardiomyopathy may result from infective, metabolic, environmental or genetic causes.5 After investigation most cases of dilated cardiomyopathy are considered to be idiopathic. Although sudden death is not uncommon in the natural history of dilated cardiomyopathy in adult life, it is unusual as a first presentation,5 so family screening in this circumstance is relatively uncommon.
Familial dilated cardiomyopathy probably accounts for up to one third or one half of idiopathic dilated cardiomyopathy.6 Genetic testing is not widely available because there is no comprehensive test for the many gene defects already identified, and because the known defects only account for a relatively small proportion of dilated cardiomyopathy known or suspected to be familial.
The diagnosis of dilated cardiomyopathy depends on clinical assessment using ECG and echocardiography. The echocardiogram is a sensitive and relatively specific non-invasive test, although defining the limit of normal ventricular size in childhood can be difficult. The sensitivity and specificity of the ECG in this circumstance are likely to be low. Most cases of familial dilated cardiomyopathy identified using echocardiography are asymptomatic.
Recommendations have been made to screen all first degree relatives of patients with dilated cardiomyopathy, even if this is not known or suspected to be familial.7 The same applies if the diagnosis of dilated cardiomyopathy is first made after sudden death. The age of onset varies greatly so echocardiographic screening of adults is recommended every 3–5 years.7 The ideal frequency of screening echocardiograms in children is not established and the age at which children should first be screened is also not established.6,7 Recognition of dilated cardiomyopathy in children as a result of family screening is uncommon. However, it seems reasonable to recommend echocardiography in all children who are first degree relatives of a victim of sudden death related to this disease and to repeat the examination every 3–5 years in childhood as well as in adult life. The benefits of early diagnosis, even in childhood, include the possibility of pre-symptomatic treatment. There is no evidence so far that earlier treatment will influence survival or the natural history and no evidence of benefit of treatment of asymptomatic gene carriers with a normal ECG and echocardiogram.
Family history of sudden death from dilated cardiomyopathy
-
Sudden death is an uncommon mode of presentation of dilated cardiomyopathy
-
Up to one third or one half of cases of dilated cardiomyopathy are familial
-
ECG and echocardiogram should be offered to all first degree relatives and repeated every 3–5 years
-
Consider genotyping in X-linked families
X-linked cardiomyopathy accounts for 5–10% of familial dilated cardiomyopathy. If this is suspected, screening should be extended to second degree relatives if the intervening relative is female. Genetic testing in X-linked families is more straightforward, with a limited number of responsible gene mutations.
MARFAN SYNDROME
Marfan syndrome is a connective tissue disorder affecting many systems including the cardiovascular system.8 It may lead to aortic root dilation and the main risk of sudden death is from aortic rupture or dissection. It is an autosomal dominant condition with a prevalence of around 1 in 5000. About one quarter of those affected represent new mutations. Marfan syndrome is a genetic disorder caused by mutations in the fibrillin-1 gene on chromosome 15. Many mutations have been identified and they are often specific to individual families. Genetic screening is complex and time consuming and at present is reserved for special situations in the UK.
Clinical diagnosis remains difficult because of phenotypic variability and variable penetrance. Diagnosis is based on a scoring system known as the Ghent criteria.8 Aortic root dilation, as detected by echocardiography, is one of the major diagnostic criteria.
No guidelines have been published for the timing or frequency of screening echocardiograms in children with a family history of Marfan syndrome. Echocardiographic assessment is not necessarily straightforward and will have to be repeated. Groenink et al reported a series of 250 children referred for screening for Marfan syndrome.8 After serial echocardiograms over a mean of five years follow up the diagnosis of Marfan syndrome was confirmed in 28%, excluded in another 42%, but remained uncertain in 30%.
The main benefit of pre-symptomatic diagnosis is detection of severe aortic root dilation to allow prophylactic aortic root replacement. Early diagnosis will also allow β blocker treatment to slow the rate of aortic root dilation and delay or prevent surgery. However, a single normal echocardiogram cannot exclude the diagnosis. In the absence of any published guidelines it seems sensible to recommend serial echocardiograms to measure the aortic root in children who have a first degree relative who has died of aortic rupture from Marfan syndrome. The frequency of such assessments is not established. A normal aortic root at maturity, in the absence of any other diagnostic criteria, probably means that further assessment in adult life is not necessary. In the long run it seems probable that advances in genetic screening tests will allow primary genetic diagnosis, but this is not likely in the near future.
Family history of sudden death from Marfan syndrome
-
Clinical examination and echocardiography in all first degree relatives
-
Repeat echocardiograms during follow up
-
Genetic testing not clinically useful at present
ARRHYTHMOGENIC RIGHT VENTRICULAR CARDIOMYOPATHY
Arrhythmogenic right ventricular cardiomyopathy or dysplasia describes a spectrum of disorders primarily affecting the right ventricle and characterised histologically by fibrous or fatty replacement of the right ventricular myocardium.10 It is associated with sudden death or ventricular tachycardia in adult life. Around 40% of arrhythmogenic right ventricular cardiomyopathy is autosomal dominant. True paediatric cases are very rare.
Diagnosis of early or mild presymptomatic cases is very difficult. An ECG and echocardiographic examination are sometimes recommended but the sensitivity and specificity are not defined in either children or adults. The most common ECG finding is T wave inversion in right precordial leads, which is normal in children. There are no guidelines for screening of children who are first degree relatives after sudden death of a family member from arrhythmogenic right ventricular cardiomyopathy.
LONG QT SYNDROME
Long QT syndrome is characterised by prolongation of the QT interval on the ECG (fig 1) and polymorphic ventricular tachycardia (torsades de pointes). It is associated with a high risk of syncope and sudden death. It is caused by an abnormality of the myocyte transmembrane sodium or potassium ion channel proteins.11 The underlying gene defects have been identified in 60–80% of patients with the long QT syndrome phenotype. They mostly have mutations of the potassium channel genes KCNQ1 (LQT1) or HERG (LQT2), while mutations in the sodium channel gene SCN5A (LQT3) are less common.11
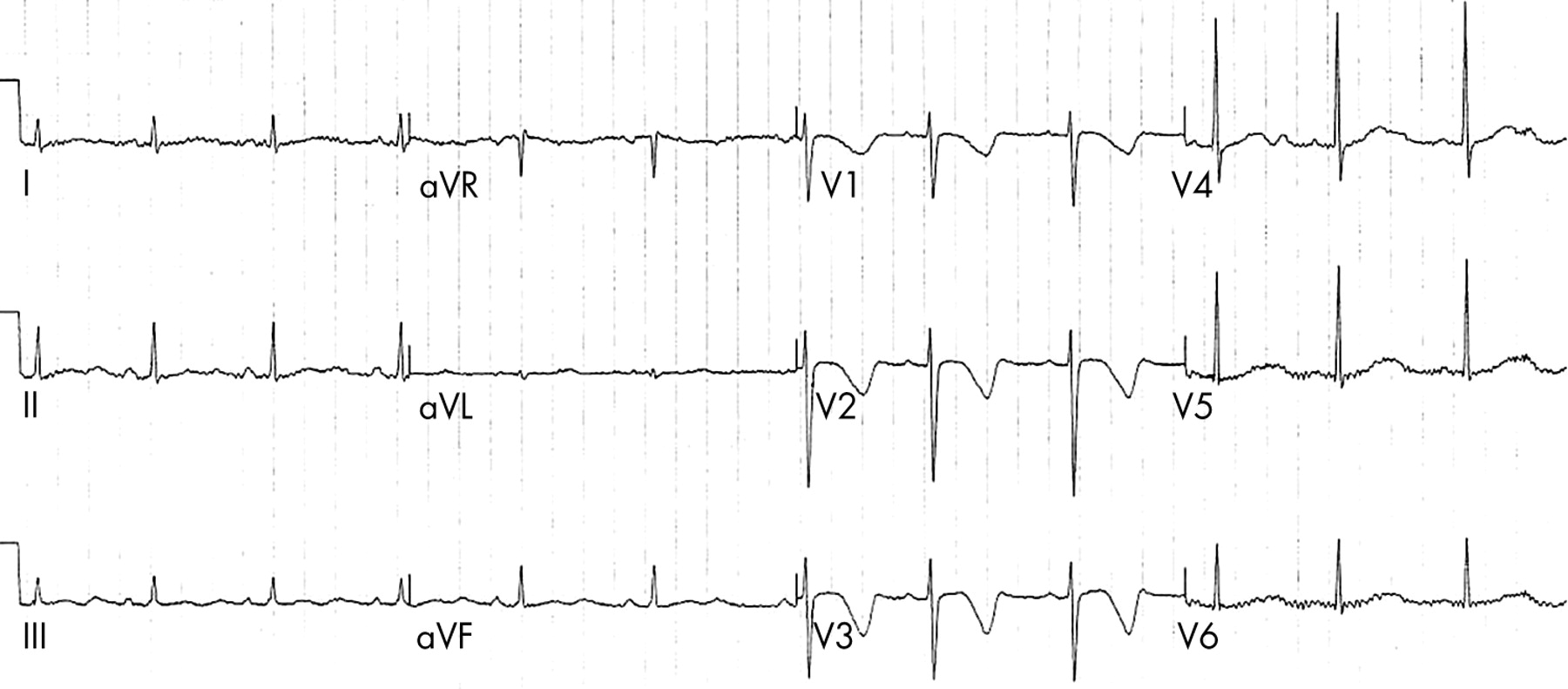
A 12 lead ECG recorded from an 11 year old girl whose sister had died suddenly with no obvious cause. The recording shows sinus rhythm with very obvious QT prolongation.
Long QT syndrome almost always has dominant inheritance although within a population there are probably more new mutations than familial cases in probands. It is very important to screen first degree relatives if long QT syndrome is identified within the family. Patients with long QT syndrome are at high risk of syncope or sudden death. The risk can be dramatically reduced by treatment with a β blocker. Recognition of long QT syndrome by genotyping or ECG screening in a young asymptomatic family member will be an indication for prophylactic β blocker treatment.
Family history of sudden death from long QT syndrome
-
ECG screening should be offered to all first degree relatives
-
ECG analysis in this circumstance is not always conclusive
-
If the genotype of the proband or other family member is known, genotyping can be extended to all first degree relatives, whether or not they have a phenotypic abnormality
-
Known gene mutations causing long QT syndrome are only found in 80% of patients with a phenotypic diagnosis
-
Identification of long QT syndrome by genotype or phenotype is an indication for treatment in the absence of symptoms
The difficulty with ECG diagnosis is the phenotypic variability. Although diagnosis can often be made with confidence from the ECG, the QT interval can be difficult to measure and has to be corrected for rate. Sometimes the rate-corrected QT measurement is not clearly normal or abnormal. The likelihood of long QT syndrome being present can be predicted from a scoring system. There is no evidence that QT analysis on Holter monitoring, exercise testing or catecholamine provocation improves diagnostic accuracy.12
Although risk is largely related to the presence of symptoms and the degree of QT prolongation, sudden death in a previously asymptomatic gene carrier who is phenotypically normal is not unknown. Because of this, it is reasonable to recommend genotyping of all first degree relatives of patients with long QT syndrome (whether or not there has been sudden death in the family).
CATECHOLAMINERGIC POLYMORPHIC VENTRICULAR TACHYCARDIA
Catecholaminergic polymorphic ventricular tachycardia is a rare cause of ventricular arrhythmia, usually recognised clinically in childhood but sometimes presenting in adult life.13 It is associated with a high risk of syncope and sudden death. The ECG at rest is usually normal and the diagnosis is made from recordings on exercise. The ECG during exercise testing, Holter monitoring or isoprenaline infusion generally shows a characteristic progression from ventricular premature beats to bidirectional ventricular tachycardia (fig 2) and polymorphic ventricular tachycardia. The condition usually has autosomal dominant inheritance and up to 20% of cases show a mutation in the cardiac ryanodine receptor gene (RyR2).11 RyR2 is the main calcium release channel on the sarcoplasmic reticulum within cardiac myocytes. Patients showing an RyR2 mutation are more likely to present in childhood and to be male compared with those with non-genotyped catecholaminergic polymorphic ventricular tachycardia. Mutations in the cardiac calsequestrin gene are described in an autosomal recessive form of catecholaminergic polymorphic ventricular tachycardia.

A 12 lead ECG showing bidirectional ventricular tachycardia characteristic of catecholaminergic polymorphic ventricular tachycardia.
β blocker treatment seems to give a high degree of protection to patients with catecholaminergic polymorphic ventricular tachycardia, but they are at great risk if the treatment is stopped or doses are missed.13 In older children and adolescents this will often be taken as an indication for an implantable defibrillator.
Genetic testing of children in the event of sudden death of a parent or sibling is generally of little help but is indicated if an RyR2 mutation has already been identified in other family members.
Because the ECG at rest is usually normal, the best way of making a clinical diagnosis is during Holter monitoring or formal exercise testing. Invasive electrophysiology studies are usually negative but the diagnosis can be confirmed with a characteristic response to isoprenaline infusion.13 If exercise or ambulatory ECG recording is negative early in childhood it may be appropriate to repeat this evaluation later in childhood as there may be age related variability in the penetrance.
Identification of asymptomatic children with ventricular arrhythmias or with RyR2 mutations will be an indication for β blocker treatment. It seems likely that many children with catecholaminergic polymorphic ventricular tachycardia have new mutations (10 of 14 (71%) in the report by Priori et al14). This is as one might expect because the natural history is to cause death before reproductive age. The implication of this finding is that the risk of siblings also having catecholaminergic polymorphic ventricular tachycardia is probably more like 15% rather than 50%.
In a recent report, Tester et al found RyR2 mutations in seven of 49 victims of sudden unexplained death with an age range of 2–34 years.15 This suggests that RyR2 mutations might be a more common explanation for unexplained sudden cardiac death than has previously been realised.
BRUGADA SYNDROME
Brugada syndrome is characterised by ECG abnormalities and a risk of sudden death from ventricular fibrillation.16 The typical ECG appearance is downsloping ST segment elevation in right precordial leads. The only treatment shown to be effective is implantation of a defibrillator. The disease mostly affects young adult males and the average age at development of arrhythmia is 40 years. Although in the original report, three of eight cases were children, diagnoses during childhood remain rare.17 Mutation of the SCN5A gene (which also causes LQT3) is found in 15–20% of patients.11,18
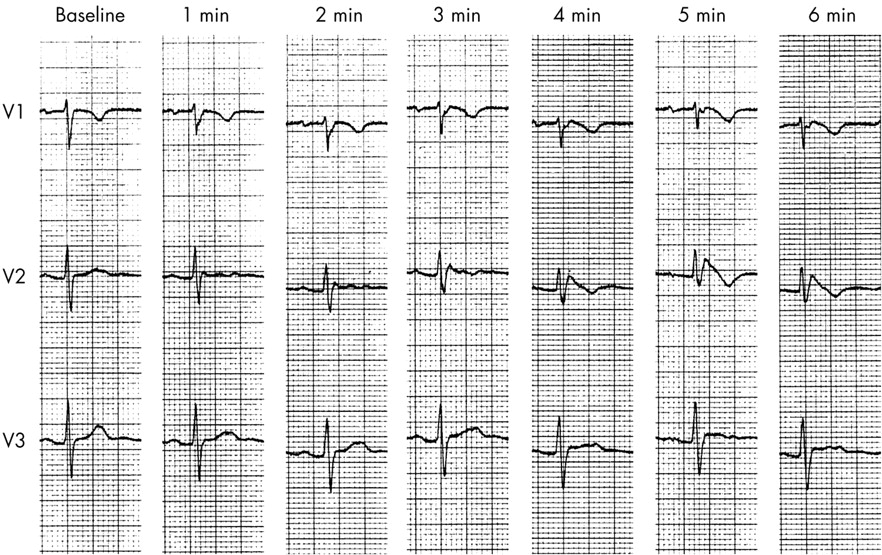
The ECG characteristics of Brugada syndrome are dynamic and variable. False positive diagnoses are known to be common in adults but the sensitivity and specificity of ECG analysis during childhood is unknown. Although there are rare case reports of children who probably have Brugada syndrome, the risk of death in childhood seems to be very low. If the diagnosis of Brugada syndrome is suspected, it can be unmasked by intravenous administration of a sodium channel-blocking drug such as flecainide (the flecainide challenge) (fig 3).19 This test has its problems as it can produce false positive results and may be negative in patients with proven Brugada syndrome. The sensitivity and specificity during childhood is unknown.
{kind=link}
{kind=link}
{kind=link}
A series of ECG recordings showing leads V1–V3 at baseline and at one minute intervals in a 15 year old girl with a family history of Brugada syndrome. Over the course of intravenous infusion of flecainide 0.8 mg/kg there is progressive development of down-sloping ST segment elevation, most obvious in lead V2.
The two main clinical difficulties are to know whether it is appropriate to test asymptomatic children with a family history of Brugada syndrome when the main risk appears in adult life; and secondly, to determine what treatment, if any, should be recommended for an asymptomatic child with an abnormal ECG or positive flecainide challenge result.
It seems that there may be some merit in a flecainide challenge in older children or young adults if Brugada syndrome is proven in a first degree relative.19 SCN5A mutation analysis is recommended in families with clinically proven Brugada syndrome, despite the relatively low mutation detection rate, since it may allow cascade testing in first degree relatives.
SUDDEN DEATH OF NO KNOWN CAUSE
The most difficult, and one of the most common, clinical situations encountered is sudden death from no known cause in a family member. This may be because the autopsy is normal in the absence of any previous history or because of the finding of coronary artery disease of uncertain significance. What screening of children in the family should be recommended in this situation? A detailed history, clinical examination, ECG and echocardiogram seem sensible recommendations for all children who are first degree relatives. The echocardiogram is likely to be unhelpful if the autopsy was normal, as that will already have excluded cardiomyopathy, Marfan syndrome, etc, as a cause of death. The ECG will be helpful to measure a normal QT interval but can be normal in some long QT gene carriers and is likely to be normal in carriers of genes for catecholaminergic polymorphic ventricular tachycardia or Brugada syndrome.
Investigation of survivors of cardiac arrest is usually extensive and may well involve invasive electrophysiology study and adrenaline provocation. This can lead to unmasking of diagnoses such as catecholaminergic polymorphic ventricular tachycardia and Brugada syndrome. However, in the absence of such a diagnosis in the family, it is not possible to recommend such aggressive investigation in asymptomatic and apparently phenotypically normal children.
Family history of sudden death from Brugada syndrome
-
Children who are first degree relatives should be offered an ECG, but the sensitivity and specificity are likely to be low
-
A flecainide challenge may be appropriate in later childhood or adolescence
-
Gene testing for SCN5A mutations can be offered if the mutation is already known to be present in the family
-
Sudden death from Brugada syndrome in childhood is probably rare
Sudden death related to swimming and thought not to be related simply to drowning seems often to be fairly specific for some sorts of cardiac channel defects. In particular, investigations have shown a high probability of the finding of LQT1 or LQT2 or CPVT1 in this situation.20 The ECG is very likely to be able to predict the presence of LQT1 or LQT2.20 In some circumstances, when the ECG is normal, it may be felt appropriate to offer genetic screening for CPVT1.20
In the past a normal autopsy after sudden death was limited to gross and histological examination. Recent recommendations include retention of tissue to allow future DNA analysis to provide the possibility of eventual confirmation or exclusion of genetic arrhythmic syndromes. At present it is not possible to recommend routine genetic analysis of children in the family for currently identified mutations in long QT syndrome, catecholaminergic polymorphic ventricular tachycardia, Brugada syndrome, etc. This is partly because the gene tests for some of these conditions are not clinically available, partly because of the cost and effort likely to be involved for a low yield, and partly because genetic analysis cannot identify all or even the majority of the underlying defects in these conditions. It is possible in the future that such analysis will be cheap, comprehensive and reliable.
CONCLUSIONS
The conditions discussed above, which may lead to sudden death with implications for the rest of the family, have an underlying genetic cause (table 1). In many situations the responsible gene defect has not been identified, or a test for it is not currently widely available for clinical use. This means that we often rely on non-invasive cardiological investigations to try to identify the phenotype. How detailed such investigations need to be depends on the primary confirmed or presumed diagnosis. Simple screening with clinical examination, ECG and echocardiography is widely available but its usefulness is limited by the variable penetrance and age-related penetrance of some of these conditions. Progress in identifying underlying genetic mutations and developing useful tests for them is rapid. It is likely that within the near future we will come to rely more on genetic analysis and less on cardiological assessment to identify those potentially at risk, although cardiological examination will continue to be an important part of risk assessment in gene carriers.
Familial causes of sudden death
Additional references appear on the Heart website—http://www.heartjnl.com/supplemental
Acknowledgments
I am indebted to Dr Paul Brennan of the Institute of Clinical Genetics, Newcastle upon Tyne for his review of the manuscript and helpful advice.
REFERENCES
Supplementary materials
Files in this Data Supplement:
- view PDF - Web-only references.
Footnotes
-
In compliance with EBAC/EACCME guidelines, all authors participating in Education in Heart have disclosed potential conflicts of interest that might cause a bias in the article. Competing interests: none.